





La poliquistosis renal autosómica dominante (PQRAD) es una enfermedad hereditaria multiorgánica, caracterizada por un progresivo crecimiento y desarrollo de quistes renales que destruyen el parénquima funcional. Es responsable del 7-10% de los casos de insuficiencia renal crónica terminal que precisan tratamiento renal sustitutivo, causada por mutaciones en los genes PKD1 y PKD2. Las dos formas de PQRAD tienen una patogenia y clínica similar, pero en los pacientes con mutación en PKD2, las manifestaciones clínicas aparecen más tarde y la progresión a nefropatía terminal acontece 10 años más tarde que en los pacientes con mutación en PKD1. El diagnóstico de esta enfermedad puede realizarse fácilmente mediante ecografía, pero el diagnóstico molecular ofrece la ventaja de la detección precoz de individuos asintomáticos portadores del defecto genético. En este trabajo, presentamos los resultados del análisis genético (PKD2) de 18 pacientes diagnosticados de PQRAD. Los objetivos de nuestro trabajo fueron comparar la rentabilidad del estudio genético respecto al radiológico, realizar un diagnóstico genético precoz en los descendientes de pacientes afectados, e intentar establecer una correlación fenotipo- genotipo en los pacientes con mutación en PKD2. Tras el análisis genético, sólo se diagnosticó a una familia (5,56 %) con mutación en el exón 13 del gen PKD2, consistente en una sustitución del nucleótido adenosina por citosina (c.2398A>C) que implicaba el cambio del aminoácido metionina por leucina (p.800Met>Leu). En nuestra población, contrariamente a lo publicado, la mutación sí se segregó con la enfermedad, y todos los miembros con diagnóstico clínico y de imagen de PQRAD presentaron dicha mutación. Dada la alta prevalencia de insuficiencia renal crónica e insuficiencia renal crónica terminal secundaria a poliquistosis renal en nuestro medio, el diagnóstico genético precoz de la poliquistosis renal conllevaría mejor pronóstico en relación con un seguimiento clínico más estricto.
INTRODUCCIÓN
La poliquistosis renal autosómica dominante (PQRAD) es una enfermedad hereditaria multiorgánica, caracterizada por el progresivo crecimiento y desarrollo de quistes renales que destruyen el parénquima funcional. Afecta aproximadamente a una de cada 1.000 personas y es responsable del 7-10% de los casos de insuficiencia renal crónica (IRC) que precisan tratamiento renal sustitutivo1.
La PQRAD está causada por mutaciones en el gen PKD1, localizado en el cromosoma 16 (16p 13.3), responsable del 85-90% de los casos, en el gen PKD2, localizado en el cromosoma 4 (4q21-23), responsable del 10-15% de los casos y, posiblemente en un tercer gen, PKD3, que aún no ha sido identificado2. Existen, además, pacientes con mutaciones en ambos genes (PKD1 y PKD2, transheterozigotos), con peor evolución clínica que los que tienen una mutación en uno solo de los genes. Las dos formas de PQRAD poseen una patogenia y una clínica similares, pero en los pacientes con mutación en PKD2 las manifestaciones clínicas aparecen más tarde y la progresión a nefropatía terminal acontece 10 años más tarde que en los pacientes con mutación en PKD1; además, los pacientes tienen una mayor esperanza de vida (69,1 frente a 53 años).
Las proteínas codificadas por los genes PKD1 y PKD2 se denominan poliquistina 1 y 2, respectivamente3,4. Son proteínas de membrana, que actúan como mecanosensores y transductores de señales, y que regulan la proliferación, adhesión, migración, diferenciación y maduración celular. Las mutaciones en los genes mencionados originan proteínas defectuosas, lo que provoca un crecimiento incontrolado del tejido y la acumulación de líquido dentro de los quistes.
La PQRAD es una enfermedad multisistémica, con manifestaciones renales y extrarrenales derivadas de la formación de quistes renales y que en muchos casos causa quistes en el hígado y páncreas5.
El diagnóstico de la PQRAD se realiza habitualmente mediante ecografía6-9. El diagnóstico genético puede utilizarse como prueba complementaria en sujetos con antecedentes familiares de PQRAD para diagnosticar de forma precoz la presencia de la enfermedad, incluso en ausencia de síntomas, en general durante la primera o la segunda década de la vida10-12, para establecer el diagnóstico definitivo de la PQRAD en pacientes con antecedentes familiares y clínica compatible con la enfermedad y en pacientes sin antecedentes de la enfermedad y clínica indicativa de PQRAD para confirmar o descartar que los quistes estén relacionados con la PQRAD. Otras ventajas ligadas al estudio genético serían: ofrecer un consejo genético con certeza en edades reproductivas, que consiste en informar al individuo afectado sobre la existencia o no de la enfermedad, su modo de herencia y los riesgos de transmitirla a su descendencia1,13,14, y contemplar la donación de órganos de familiares de los pacientes afectados.
El diagnóstico molecular no puede predecir el momento de comienzo, la gravedad, el tipo de síntomas o el grado de progresión de la enfermedad; sin embargo, el diagnóstico precoz de la PQRAD conllevaría un mejor pronóstico, al permitir un seguimiento clínico más estricto15.
El diagnóstico genético puede realizarse por búsqueda directa de la mutación o de forma indirecta, mediante análisis de ligamiento16-19. El análisis mutacional presenta dificultades debido al gran tamaño y complejidad del gen PKD1, y a la gran cantidad de mutaciones y polimorfismos descritos en dicho gen, que hace difícil distinguir los cambios patogénicos de los neutrales. Una opción diagnóstica en estos pacientes sería, por tanto, el análisis de ligamiento genético, para lo que se requieren varios miembros de la familia afectados.
En nuestro estudio, nos planteamos, dada la dificultad para el diagnóstico genético de pacientes con PQRAD y mutación en el gen PKD1 y, a pesar de su mayor prevalencia, realizar el análisis mutacional del gen PKD2 en los pacientes vivos no emparentados con diagnóstico clínico y radiológico de PQRAD, con el objetivo de comparar la rentabilidad del estudio genético respecto al radiológico, realizar un diagnóstico precoz de PQRAD en los descendientes de pacientes afectados y, por último, intentar establecer correlación fenotipo-genotipo en los pacientes con mutación en PKD2.
MATERIAL Y MÉTODOS
Población estudiada
Hemos realizado el estudio genético en 18 de los 48 pacientes con PQRAD, que precisaron atención médica en el Servicio de Nefrología del Hospital Universitario de Salamanca durante el período 1994-2005 y seguimiento hasta abril de 2008; un miembro de cada familia en la que existía certeza del carácter hereditario de la enfermedad. En los pacientes en los que se demostró mutación en el gen PKD2, se extrajeron muestras de los familiares vivos. Los pacientes habían sido diagnosticados de PQRAD de acuerdo con criterios clínicos y radiológicos. Las muestras fueron obtenidas previo consentimiento informado.
Estudio genético
Se extrajeron 10 ml de sangre periférica por venopunción, a partir del cual se obtuvo el ADN, para el estudio molecular.
Posteriormente se amplificaron los exones del gen PKD2 por PCR. No se amplificó el exón 1 debido a las dificultades encontradas en la amplificación de la PCR por la riqueza en residuos de guanina y citosina.
Una vez amplificados, los fragmentos fueron analizados mediante la técnica de CSGE-heterodúplex para determinar la presencia de alguna mutación en la región codificante del gen (figura 1).
Todos los productos de PCR en los que se detectaron variantes fueron secuenciados.
Se utilizaron el programa bioinformático «Nnpredict» para comprobar si las mutaciones encontradas eran o no responsables de cambios estructurales dentro de la proteína y, el programa «Esefinder» para comprobar si las mutaciones residían o no en regiones exónicas que regularan el procesamiento del ARN.
Seguimiento clínico
Se analizaron los principales aspectos clínicos de los pacientes y familiares con PQRAD en los que se demostró la existencia de una mutación en el gen PKD2. La información de todos los datos se obtuvo de las historias clínicas. Los principales parámetros analizados fueron edad de aparición de la enfermedad, aspectos relacionados con las manifestaciones clínicas iniciales de la enfermedad, con las manifestaciones clínicas aparecidas en la evolución, con las manifestaciones extrarrenales y con la supervivencia y morbimortalidad.
Las variables cualitativas se expresaron como porcentajes y las variables numéricas como media y desviación estándar (X ± DE).
RESULTADOS
Análisis genético
Se realizó el estudio genético en busca de mutaciones en PKD2, en 18 pacientes, 9 mujeres y 9 hombres. Cada uno de ellos pertenecía a una de las familias en las que había certeza del carácter hereditario de la PQRAD.
El único hallazgo fue encontrado en un hombre, en quien se halló una sustitución del nucleótido adenosina por citosina en posición 2398 (c.2398 A>C) (figura 2), que implicaba el cambio del aminoácido metionina (Met) por leucina (Leu) en la posición 800 (p.800 Met>Leu).
El estudio de la poliquistina 2 mutada por métodos bioinformáticas no detectó cambios estructurales en la proteína.
La mutación no residía en una región exónica que regulara el procesamiento del ARN (figura 3).
Realizado el árbol genealógico y extraídas muestras de sangre de los familiares vivos se confirmó el mismo hallazgo en sus 2 hermanas, las cuales tenían afectación clínica pero no en sus hijas, que estaban sanas (figura 4).
Análisis clínico de pacientes con mutación en PKD2
Realizado el árbol genealógico de los pacientes portadores de la mutación en el gen PKD2, se confirmó el carácter hereditario de la enfermedad (figura 4). La transmisión había sido por vía paterna y también había constancia de la enfermedad en el abuelo paterno (figura 4), ambos fallecidos.
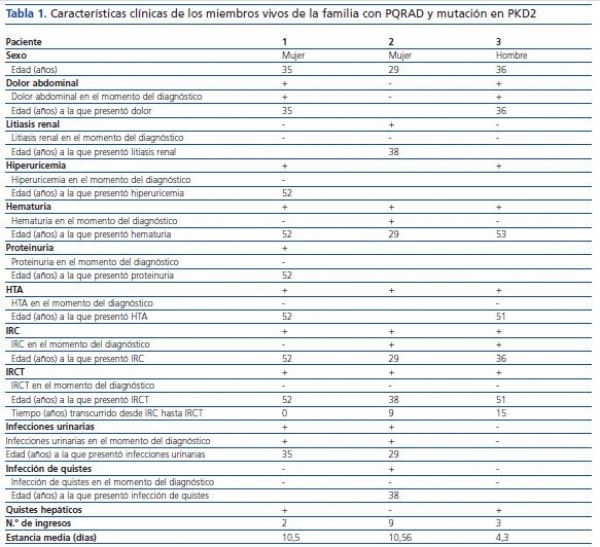
De los 3 pacientes afectados vivos, 2 eran mujeres y uno era un hombre. La edad media del diagnóstico fue 33 ± 3,79 años (tabla 1). La insuficiencia renal, presente en el 100 % de los portadores de la mutación, fue síntoma de presentación de la enfermedad en dos de ellos; otros síntomas presentes en el momento del diagnóstico fueron el dolor abdominal (en dos de ellos), hematuria macroscópica (un paciente) y las infecciones del tracto urinario (2 pacientes). Con el transcurso de la enfermedad desarrollaron litiasis renal (uno de ellos), hiperuricemia (2 pacientes), hematuria presente en el 100% con una edad media de aparición de 51,5 ± 0,71 años, HTA en el 100%, e IRCT en el 100%, con una edad media de aparición de 35,25 ± 24,35 años y un tiempo transcurrido desde el diagnóstico de la IRC de 4,8 ± 6,91 años. Como única manifestación extrarrenal, 2 de los 3 pacientes afectados presentaron quistes hepáticos en el momento del diagnóstico detectados mediante ecografía (tabla 1).
Realizado el estudio clínico y genético de los descendientes se descartó la existencia de PQRAD en ellos.
DISCUSIÓN
Diagnóstico genético
Mediante el empleo de técnicas diagnósticas de imagen en sujetos con antecedentes familiares de PQRAD es posible diagnosticar de forma precoz la enfermedad, incluso en ausencia de síntomas, generalmente durante la primera o la segunda décadas de la vida6-9,16-18. Sin embargo, en cerca del 40% de los pacientes no se reconocen antecedentes familiares, lo que sugiere bien una alta tasa de mutación de novo, o bien la existencia de genes modificadores que afecten a la expresión de la enfermedad20, lo que dificulta el diagnóstico precoz en estos pacientes. Una alternativa diagnóstica cada vez más empleada es el diagnóstico genético.
Las dos formas de PQRAD tienen una patogenia y clínica similar; aunque en la PQRAD con mutación en PKD2, las manifestaciones clínicas y la progresión a nefropatía terminal acontecen más tarde y los pacientes tienen una mayor esperanza de vida21, no existen rasgos fenotípicos que nos permitan diferenciar en el momento del diagnóstico una de otra. De ahí la importancia de recurrir al estudio genético como herramienta para el diagnóstico precoz, especialmente en familias con mutación en el gen PKD2, en las que el análisis genético tiene más sensibilidad que el estudio ecográfico, sobre todo en las primeras décadas de la vida, y permite diagnosticar la alteración antes de que se desarrollen quistes renales y aparezcan síntomas clínicos16.
En nuestro estudio, la prevalencia de pacientes con PQRAD y diagnóstico de mutaciones en PKD2 fue inferior a la descrita (5,56 frente a 10-15%)22,23, lo que nos induce a pensar en una mayor prevalencia de pacientes con mutación en PKD1, lo que concordaría con la evolución clínica hacia la IRCT precoz de los pacientes estudiados, o en una selección sesgada de los pacientes a los que se le realizó el estudio genético. Dado que la PQRAD con mutación en PKD2 se manifiesta más tarde clínicamente y sólo analizamos a los pacientes en los que existía certeza del carácter hereditario de la enfermedad, pudo ocurrir que no analizáramos genéticamente a pacientes con PQRAD y progenitores con mutación en el gen PKD2 no diagnosticados de PQRAD por presentar manifestaciones clínicas leves de la enfermedad o por fallecer precozmente sin haber diagnosticado la enfermedad.
Dada la alta prevalencia de IRC y de insuficiencia renal crónica terminal (IRCT) secundaria a PQRAD en nuestro estudio, el diagnóstico precoz de la PQRAD conllevaría un mejor pronóstico en relación con un seguimiento clínico más estricto. En este sentido, el diagnóstico molecular ofrece la posibilidad de una intervención precoz en lo que se refiere al seguimiento y tratamiento de la HTA, de infecciones o litiasis y, como consecuencia, retrasar la aparición de la IRC, disminuir la incidencia de pacientes con necesidad de tratamiento renal sustitutivo, y la comorbilidad que puede agravar la evolución hacia la insuficiencia renal y secundariamente la mortalidad. Un tema de interés en relación con el diagnóstico precoz es tener un seguimiento de estos pacientes, saber si existe una tendencia en determinadas familias o individuos a manifestarse precozmente, y si los síntomas iniciales tienen un efecto adverso en la evolución de la enfermedad.
Análisis de la mutación y segregación familiar
En nuestro estudio, tras el análisis genético, se diagnosticó a una familia con mutación en el exón 13 del gen PKD2 en la que se observó una sustitución del nucleótido adenosina por citosina en posición 2398 (c.2398 A>C) que implicaba el cambio del aminoácido Met por Leu en la posición 800 (p.800 Met>Leu).
El significado de dicha mutación es indeterminado y, aunque ya ha sido descrita por otros grupos que observan que dicha mutación no se segrega con la enfermedad10,20, nosotros, por el contrario, encontramos que todos los miembros con diagnóstico clínico y de imagen de PQRAD presentaron dicha mutación; por lo tanto, discrepamos con los estudios referidos y confirmamos que en nuestro estudio la mutación sí se segregó con la enfermedad.
La poliquistina 1 y la poliquistina 2 interaccionan a través del extremo C terminal24,25, mediado por el dominio coiled-coil de la poliquistina 2 situado, según las diferentes publicaciones, en el exón 12 y 13 de la poliquistina 226 o en la región de la poliquistina 2 comprendida entre el codón 872 y el extremo carboxiterminal (exones 14 y 15)27. Parece ser que los pacientes con mutación en el extremo 3’ presentan menos complicaciones renales (HTA, hematuria, litiasis renal, infecciones urinarias, etc.), ya que la poliquistina mutada puede conservar la función28. Estos datos, referidos en la literatura, nos conducen a pensar que la mutación encontrada en nuestra población tendría escasa significación clínica. El estudio estructural de la proteína por métodos bioinformáticos confirmó la ausencia de significación estructural, probablemente porque dicha mutación afecta a un dominio no importante en la estructura de la proteína. El análisis de la región exónica donde se encontraba la mutación determinó que dicha región no regulaba el procesamiento del ARN.
En nuestros pacientes la mutación tuvo significación clínica y todos los pacientes con mutación en el exón 13 se comportaron clínicamente como pacientes con mutación en el gen PKD1 y evolucionaron rápidamente hacia la insuficiencia renal. Esto nos hace plantearnos que los miembros de la familia afectada puedan presentar, además, una mutación en el gen PKD1 (transheterocigotos), lo que explicaría el curso clímás grave de la enfermedad, no justificado por una mutación aislada ni en el gen PKD1 ni en el gen PKD227. La ausencia de manifestaciones clínicas y genéticas en la descendencia no excluye totalmente la enfermedad, ya que el diagnóstico por técnicas de imagen en la tercera década de la vida no ofrece resultados concluyentes y sería preciso llevar a cabo en posteriores investigaciones el análisis del gen PKD1 para excluir la enfermedad en los descendientes, sanos en el momento del estudio.
En nuestro trabajo no pudimos confirmar ningún tipo de correlación clínico-genética, resultado no concluyente dado el escaso número de pacientes afectados por la mutación en el gen PKD2, y el patrón evolutivo entre los miembros afectados fue muy diferente. Esta heterogeneidad puede atribuirse a la existencia de genes modificadores que aumenten la gravedad del defecto20.
Dada la alta prevalencia de pacientes con PQRAD diagnosticados tardíamente, en estadios avanzados de IRC e IRCT, es preciso protocolizar el diagnóstico genético precoz como forma de reducir las complicaciones asociadas con la enfermedad. En resumen, las mutaciones en el gen PKD2 son una causa minoritaria de PQRAD en nuestro medio, por lo que consideramos de gran importancia analizar el gen PKD1 para poder llevar a cabo un diagnóstico genético en estos pacientes.

Figura 1.

Figura 2.

Figura 3.

Figura 4.

Tabla 1.



