



Introducción: Las alteraciones en el diafragma de filtración y/o citoesqueleto del podocito están relacionadas con proteinuria y síndrome nefrótico. En nuestra población, la glomerulopatía más frecuente, demostrada por biopsia, es la glomerulosclerosis focal y segmentaria. Nuestro objetivo fue buscar alteraciones en la expresión de algunas de las proteínas asociadas con el diafragma de filtración en pacientes con proteinuria en rango nefrótico. Métodos: Tejido renal de 40 pacientes con proteinuria en rango nefrótico, de 10 pacientes con proteinuria leve, de tres con hematuria aislada y 10 muestras de tejido renal normal (donantes cadáveres) se estudiaron, por inmunofluorescencia indirecta, para expresión de nefrina, podocina y α-actinina 4. Resultados: La expresión de estas proteínas fue lineal, homogénea, en las paredes capilares glomerulares del tejido renal normal y de pacientes con hematuria aislada. En proteinuria nefrótica y en algunos casos de proteinuria leve este aspecto normal estaba alterado y su expresión cambió de lineal a granular fina. En 22 casos (45%) de pacientes con proteinuria nefrótica y en 3 casos (30%) de pacientes con proteinuria subnefrótica hubo pérdida en la expresión de al menos una de estas proteínas (p = 0,49). Estas alteraciones se encontraron en las diferentes glomerulopatías que de forma más habitual causan síndrome nefrótico, aunque ninguna en particular fue significativamente más frecuente. Conclusiones: En la proteinuria nefrótica es muy frecuente la redistribución o la pérdida de proteínas asociadas al diafragma de filtración, lo que en muchos casos podría ser una consecuencia más que una causa de la proteinuria. Estas alteraciones pueden evidenciarse también en pacientes con proteinuria leve.
INTRODUCCIÓN
Entre las causas de síndrome nefrótico la enfermedad glomerular de cambios mínimos, la glomerulonefritis membranosa y la glomerulosclerosis focal y segmentaria (GEFyS) son las glomerulopatías más frecuentes1-5. Esta última es también la enfermedad glomerular más frecuente, diagnosticada por biopsia, en Colombia6,7.
Las células epiteliales viscerales del glomérulo están unidas a la membrana basal glomerular por medio de extensiones citoplasmáticas que forman interdigitaciones conocidas como procesos podocitarios8. Normalmente el filtrado glomerular pasa entre estos procesos podocitarios a través de una unión intercelular especializada, exclusiva de podocitos, que es conocida como «hendidura de filtración» (o «diafragma de hendidura », slit diaphragm), que aquí llamaremos, por ser un término que describe mejor su estructura y función, «diafragma de filtración». Esta estructura está anclada al citoesqueleto de los procesos podocitarios, y en este complejo hay numerosas proteínas estructurales y con funciones de mensajeros que no han sido completamente conocidas ni entendidas9,10. Hasta el momento se han caracterizado al menos 15 proteínas que forman parte de este complejo, todas ellas importantes en el mantenimiento de su estructura y función: nefrina, podocina, NEPH1, 2 y 3, P-cadherina, proteína asociada a CD2 (CD2AP), cateninas, FAT 1 y 2, zonula occludens 1 (ZO-1), actina, alfa-actinina-4, densina y CRIM19,11-21. Desde finales de los noventa, con el descubrimiento de varias mutaciones en genes que codifican estas moléculas, se descubrió que ésta es la principal barrera para la filtración de proteínas9,11,12,22.
En respuesta a muchos tipos de agresión, los podocitos sufren un cambio morfológico denominado «borramiento» (o fusión) de procesos podocitarios, que se debe a una alteración arquitectural del citoesqueleto y la unión intercelular8. Por microscopia electrónica, este borramiento se manifiesta con retracción y franca ampliación de las extensiones citoplasmáticas de podocitos. Este proceso es reversible y está directamente relacionado con proteinuria en humanos y modelos experimentales23.
Estudios moleculares indican que las proteínas del diafragma de filtración forman una estructura similar a una cremallera con una periodicidad constante de 40 nm9,24,25. La localización exacta y las interacciones precisas entre las proteínas del diafragma de filtración no se conocen completamente. Estas proteínas interactúan con el citoesqueleto del podocito y participan en la transmisión de señales intracelulares e intercelulares. Parece ser que el diafragma de filtración, además de ser un filtro dependiente del tamaño de las moléculas, también ejerce su función de acuerdo con la carga eléctrica, pudiendo así repeler las proteínas y evitando que este diafragma se tapone26.
Cuando hay alteraciones en alguno de los genes que codifican estas proteínas, todo el complejo puede perder su estructura normal y alterar su función llevando a un síndrome nefrótico, usualmente congénito. Sin embargo, en casos de síndrome nefrótico adquirido, mediado o no por complejos inmunitarios y/o inmunidad celular, se describen alteraciones podocitarias (fusión o borramiento de procesos podocitarios) como un fenómeno secundario al síndrome nefrótico (o a su causa)9,24. Por lo tanto, parece ser que la desestructuración y la pérdida de función del diafragma de filtración y su complejo anclaje de proteínas pueden ser tanto causa como consecuencia del síndrome nefrótico.
Siendo la GEFyS la glomerulopatía más frecuente, diagnosticada por biopsia, en nuestra población6,7, es muy importante buscar mecanismos moleculares relacionados con su desarrollo para así entender mejor su etiología. Nuestro objetivo fue buscar la alteración en la expresión de algunas de estas proteínas en paciente nefróticos de Colombia.
MATERIAL Y MÉTODOS
Casos de biopsias renales de pacientes con proteinuria en rango nefrótico (n = 40), en los que se dispuso de tejido fresco congelado tras el estudio convencional para inmunofluorescencia (IF), fueron seleccionados detectar algunas de las proteínas asociadas con el diafragma de filtración: nefrina, extremo C-terminal (C-ter), podocina, extremo C-ter, y α-actinina 4, extremo C-ter. Como controles normales se utilizaron 11 casos de tejido renal normal proveniente de donantes renales cadáveres, con estudio microscópico demostrando histología normal, IF negativa para depósitos de inmunoglobulinas y/o fracciones del complemento, y pruebas de función renal y citoquímico de orina normales. Para efectos de comparación se incluyeron también 10 casos de pacientes con proteinuria no nefrótica y 3 casos de pacientes con hematuria aislada (sin proteinuria ni otras alteraciones de la función renal).
Las muestras provienen de pacientes con edades entre 1 y 71 años (mediana: 25); el 59,4% fueron hombres. La mediana de la edad de los pacientes con proteinuria nefrótica fue de 13,5 años (1-71); la mediana de la edad de los pacientes con proteinuria no nefrótica fue de 33 años (9-67); la mediana de la edad de pacientes con hematuria aislada fue de 11 años (6-52), y la mediana de la edad de los donantes cadáveres con tejido renal normal fue de 48 años (20-60).
Inmunofluorescencia
Las biopsias renales fueron procesadas de acuerdo a nuestro protocolo de procesamiento sistemático, basado en las recomendaciones internacionales27. Tejido fijado en formalina e incluido en parafina fue utilizado para el estudio de microscopia de luz convencional. Para IF directa (para detectar IgA, IgG, IgM, C3, C1q, y cadenas ligeras kappa y lambda), las muestras fueron congeladas inmediatamente usando gel adecuado para cortes por congelación (Tissue-Tek, Nile, Inc., Eckhart, IN, EE.UU.); se realizaron los cortes necesarios para la IF diagnóstica y del tejido restante se realizaron cortes adicionales de 3 μm de espesor, en placa cargada, adecuada para técnicas de inmunohistoquímica (Fisher Scientific, EE.UU.). Estos cortes fueron fijados en acetona, a temperatura ambiente, durante 10 minutos y luego fueron almacenados a -24 ºC hasta su utilización para la detección de proteínas asociadas con el diafragma de filtración. En todos los casos, antes de los cortes para el presente estudio, se confirmó la presencia de, al menos, 5 glomérulos.
Para la detección de proteínas asociadas con el diafragma de filtración utilizamos la técnica de IF indirecta. Todos los anticuerpos primarios y secundarios, estos últimos marcados con fluoresceína isoticianato (FITC), son policlonales y fabricados por el Laboratorio Santacruz Biotechnology Inc. (EE.UU.): nefrina: número de catálogo sc32530, podocina: número de catálogo sc22296, α-actinina 4: número de catálogo: sc7454, anticuerpo secundario: anti-IgG de cabra proveniente de burro y marcado con FITC: número de catálogo: sc2024, suero normal de burro: número de catálogo: sc2044.
Los pasos de la IF fueron los siguientes: los cortes fueron descongelados, lavados con tris-buffer fosfato (TBS): dos lavados de 5 minutos, incubados con suero normal (para bloquear unión inespecífica) derivado de la misma especie de la cual se origina el anticuerpo secundario, al 10% (dilución 1:10) durante 20 minutos, lavados con TBS durante 5 minutos, incubados con el anticuerpo primario durante 60 minutos, dilución 1:20, lavados tres veces, durante 5 minutos, con TBS, incubados durante 45 minutos con el anticuerpo secundario conjugado con la FITC, dilución: 1:20, lavados tres veces, durante 5 minutos, con TBS, las placas histológicas fueron finalmente montadas con glicerina al 90% y almacenadas a 4 ºC, en ausencia de luz, hasta su observación con el microscopio de fluorescencia.
Interpretación
La inmunotinción fue evaluada en las paredes de capilares glomerulares y su intensidad fue evaluada subjetivamente por comparación con los controles de tejido renal normal; se registró el patrón de inmunotinción: lineal o granular, y homogéneo o irregular (heterogéneo en diferentes áreas del glomérulo). Se analizaron imágenes estáticas de todos los casos positivos con Phothoshop®, versión 10, para determinar la relación de la inmunotinción con la membrana basal glomerular.
Análisis estadístico
Los datos son expresados como mediana (y valores mínimomáximo). Para comparar porcentajes utilizamos las pruebas de la chi-cuadrado o de Fisher, según el número de casos y sus valores. Los análisis se relaizaron con el software SPSS®, Chicago, IL, versión 15.
RESULTADOS
De los 40 pacientes con proteinuria en rango nefrótico, 37 tenían síndrome nefrótico completo. La biopsia renal confirmó el diagnóstico de GEFyS en 15 de estos casos, enfermedad de cambios mínimos en siete, glomerulonefritis (GN) membranosa en ocho, nefropatía IgA en dos, GN postinfecciosa en dos, y un caso cada uno de GN membranoproliferativa tipo I, enfermedad de depósitos densos, nefropatía diabética, amiloidosis, nefropatía asociada con el virus de la inmunodeficiencia humana (VIH) y nefropatía C1q.
En los 10 casos de pacientes con proteinuria en rango no nefrótico el diagnóstico fue nefropatía IgA en 6 casos y uno cada uno con GEFyS, nefropatía IgM, nefropatía hipertensiva y GN proliferativa mesangial no IgA. Tres biopsias de pacientes con hematuria aislada, cuyos diagnósticos en la biopsia renal fueron: nefropatía IgA, enfermedad de membranas basales delgadas y GN proliferativa mesangial no IgA, fueron también incluidos.
Expresión de proteínas asociadas con el diafragma de filtración
Biopsias normales
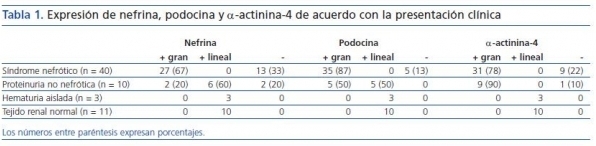
En los 11 riñones normales los anticuerpos policlonales contra la podocina, nefrina y α-actinina-4 presentan el mismo patrón de inmunomarcación: positividad lineal, homogénea, difusa, en paredes capilares glomerulares periféricas (figura 1), en algunas zonas se evidencia una localización a lo largo de la porción externa de la membrana basal glomerular. Esta inmunotinción será considerada en adelante como «normal».
Biopsias de pacientes con proteinuria nefrótica
Hubo más casos con pérdida de la expresión de nefrina, podocina y α-actinina-4 en pacientes con síndrome nefrótico que en pacientes con proteinuria subnefrótica, aunque la diferencia no fue estadísticamente significativa (p = 0,70, p = 0,57 y p = 0,66, respectivamente) (tabla 1). La expresión de estas proteínas fue granular, con gránulos finos o gruesos en la parte externa de los capilares glomerulares (figura 2), en la mayoría de casos difusa; sólo en algunos casos, con lesiones hialinas o esclerosantes segmentarias, esta positividad fue irregular y/o segmentaria. En ninguno de los casos de proteinuria en rango nefrótico la expresión de estas proteínas fue lineal.
Biopsias de pacientes con proteinuria no nefrótica
La expresión fue variable, con algunos casos de pérdida de la expresión de nefrina o de α-actinina-4. No hubo casos con pérdida de la expresión de podocina. En cuanto al patrón de expresión (lineal o granular), éste fue variable, con algunos casos lineal y otros con aspecto granular similar al encontrado en casos con proteinuria nefrótica (tabla 1).
Biopsias de pacientes con hematuria aislada
En los 3 casos la inmunotinción para nefrina, podocina y α-actinina-4 fue lineal, similar a la del tejido renal normal. No hubo ningún caso con pérdida de la expresión de alguna de estas proteínas.
En 2 casos hubo pérdida simultánea en la expresión de las tres proteínas estudiadas; uno de estos casos corresponde a enfermedad de depósitos densos y el otro a nefropatía asociada con el VIH. En 5 casos hubo pérdida simultánea en la expresión de dos de estas proteínas, todos ellos en pacientes con proteinuria nefrótica, y en 14 casos hubo pérdida de la expresión de sólo una de las proteínas estudiadas, 11 en pacientes con síndrome nefrótico y tres en pacientes con proteinuria no nefrótica. En total en 18 de los 40 casos (45%) de pacientes con proteinuria en rango nefrótico hubo pérdida de la expresión de al menos una de las proteínas estudiadas, y en 3 casos de proteinuria no nefrótica (30%) hubo pérdida en la expresión de una de ellas (p = 0,49). En ninguna de las muestras provenientes de pacientes con proteinuria no nefrótica hubo pérdida simultánea en la expresión de varias proteínas.
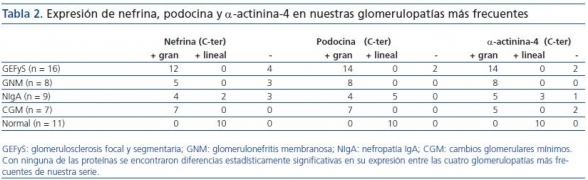
Con respecto a la expresión de estas proteínas en diferentes tipos histológicos de glomerulopatías, no encontramos diferencias estadísticamente significativas, aunque es llamativo que en enfermedad glomerular de cambios mínimos no se produjeron casos de pérdida de expresión de nefrina o de podocina, y sólo 2 de 7 casos mostraron pérdida de la expresión de α-actinina-4 (tabla 2).
La intensidad de la inmunotinción varió algo entre casos, pero esta variación es completamente subjetiva ya que no disponemos de un sistema de cuantificación de la intensidad; además, ésta puede variar de acuerdo con la exposición a la luz de cada campo microscópico debido a la pérdida progresiva de la fluorescencia con la fotoexposición. En lesiones esclerosantes segmentarias fue frecuente encontrar una pérdida de la expresión de las proteínas asociadas con el diafragma de filtración. En ninguna de las muestras estudiadas detectamos expresión de alguna de estas proteínas en sitios extraglomerulares.
DISCUSIÓN
Alteraciones genéticas en alguna de las proteínas que hacen parte del diafragma de filtración se han asociado con proteinuria o con síndrome nefrótico de inicio temprano. Sin embargo, otras causas no genéticas de síndrome nefrótico producen también alteraciones de aquéllas9,22,28. En el presente trabajo nosotros intentamos inicialmente conocer si hay alguna alteración más frecuente en nuestros pacientes con síndrome nefrótico; sin embargo, no encontramos alguna alteración característica o asociada con mayor frecuencia a alguna de las glomerulopatías, entre ellas la GEFyS, la cual es la enfermedad glomerular más frecuente en nuestros pacientes.
En nuestro trabajo, encontramos que en casos de proteinuria en rango nefrótico, independientemente de su causa, hay alteración en la expresión de la nefrina, la podocina y la α-actinina-4. Esta alteración también se evidencia en algunos casos de proteinuria leve (no nefrótica), aunque con menor frecuencia. La pérdida de la expresión de algunas de estas proteínas es un episodio común en casos de proteinuria en rango nefrótico; sin embargo, esto no indica que haya mutaciones en los genes que las codifican; la pérdida simultánea de varias de estas proteínas en algunos de nuestros casos indica una alteración no genética.
En riñones normales la nefrina, podocina y α-actinina-4 aparecen codistribuidas a lo largo de la parte externa de la membrana basal glomerular. Esta distribución normal se pierde en casos de proteinuria nefrótica, lo que sugiere que la alteración podocitaria encontrada en estos pacientes tiene relación directa con la redistribución o pérdida de una o varias de las proteínas asociadas con el diafragma de filtración. En casos de mutación de genes codificando alguno de los extremos de estas proteínas podría haber una pérdida en la expresión de dicho extremo, pero puede conservarse la inmunomarcación del otro extremo29, o la proteína mutada se continúa detectando, pero estaría alterada su localización30. Esto indica que en nuestros casos es posible que aunque se conserve la inmunotinción para uno de los extremos de la nefrina, la podocina o la α-actinina 4, este hallazgo no descarta la presencia de mutaciones en los genes que las codifican. Nosotros intentamos hacer inmunotinción de los otros extremos (N-ter) de la nefrina, de la podocina y de α-actinina-4, pero no logramos resultados técnicamente adecuados con los anticuerpos utilizados (del mismo fabricante) y, por lo tanto, los resultados no se presentan. De igual manera, intentamos determinar la alteración en diferentes porciones de las proteínas CD2AP, TRPC6 y NEPH1, sin lograr resultados satisfactorios con la técnica. El estudio de estas otras proteínas en los casos del presente trabajo sería muy útil para conocer otras alteraciones o para determinar la frecuencia de pérdidas simultáneas de otras moléculas.
La expresión de la nefrina, podocina y α-actinina-4 como granular y no lineal indica que estas proteínas se han redistribuido en el citoplasma podocitario y no logran ensamblarse como en la estructura normal de glomérulos sin enfermedad; sin embargo, nuestros resultados indican que esto puede ser una consecuencia del síndrome nefrótico de diversa etiología, y no necesariamente una causa. Al parecer, los podocitos responden, de alguna manera, a la proteinuria y alteran el ensamblaje de estas proteínas; en consecuencia, estas proteínas representan una diana terapéutica en la proteinuria, como parece sugerir la modificación de la proteinuria con el uso de los inhibidores de la enzima de conversión de la angiotensina (IECA) o de los antagonistas de receptores de angiotensina II (ARA II) en algunos pacientes, en los que se demuestra modificación en la expresión de nefrina20,21.
Estos hallazgos plantean varios puntos muy importantes en la patogenia de la proteinuria: 1) en proteinuria grave (rango nefrótico), hay desestructuración del diafragma de filtración, lo cual puede ser causa y/o consecuencia de la proteinuria; 2) además de la redistribución de estas proteínas, es frecuente la pérdida de la expresión de algunas de ellas (o de alguna porción en particular), sin que necesariamente esto implique alteraciones genéticas; 3) en proteinuria en rango no nefrótico, aunque menos frecuente, pueden evidenciarse alteraciones en la expresión de proteínas asociadas al diafragma de filtración, similares a las detectadas en el síndrome nefrótico, y 4) estas alteraciones se relacionan con proteinuria y no se evidencian en casos de hematuria aislada.
En conclusión, en pacientes con proteinuria nefrótica es muy frecuente la alteración (redistribución o pérdida) de proteínas asociadas con el diafragma de filtración, lo que en muchos casos parece ser una consecuencia más que una causa de la proteinuria. Estas alteraciones pueden evidenciarse también, aunque con menor frecuencia, en pacientes con proteinuria subnefrótica.

Figura 1.

Figura 2.

Tabla 1.

Tabla 2.



