





La poliquistosis renal autosómica dominante (PQRAD) es una enfermedad hereditaria multiorgánica, caracterizada por un progresivo crecimiento y desarrollo de quistes renales que destruyen el parénquima funcional. Es responsable del 7-10% de los casos de insuficiencia renal crónica terminal que precisan tratamiento renal sustitutivo, causada por mutaciones en los genes PKD1 y PKD2. Las dos formas de PQRAD tienen una patogenia y clínica similar, pero en los pacientes con mutación en PKD2, las manifestaciones clínicas aparecen más tarde y la progresión a nefropatía terminal acontece 10 años más tarde que en los pacientes con mutación en PKD1. El diagnóstico de esta enfermedad puede realizarse fácilmente mediante ecografía, pero el diagnóstico molecular ofrece la ventaja de la detección precoz de individuos asintomáticos portadores del defecto genético. En este trabajo, presentamos los resultados del análisis genético (PKD2) de 18 pacientes diagnosticados de PQRAD. Los objetivos de nuestro trabajo fueron comparar la rentabilidad del estudio genético respecto al radiológico, realizar un diagnóstico genético precoz en los descendientes de pacientes afectados, e intentar establecer una correlación fenotipo- genotipo en los pacientes con mutación en PKD2. Tras el análisis genético, sólo se diagnosticó a una familia (5,56 %) con mutación en el exón 13 del gen PKD2, consistente en una sustitución del nucleótido adenosina por citosina (c.2398A>C) que implicaba el cambio del aminoácido metionina por leucina (p.800Met>Leu). En nuestra población, contrariamente a lo publicado, la mutación sí se segregó con la enfermedad, y todos los miembros con diagnóstico clínico y de imagen de PQRAD presentaron dicha mutación. Dada la alta prevalencia de insuficiencia renal crónica e insuficiencia renal crónica terminal secundaria a poliquistosis renal en nuestro medio, el diagnóstico genético precoz de la poliquistosis renal conllevaría mejor pronóstico en relación con un seguimiento clínico más estricto.
INTRODUCTION
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is a hereditary multi-organic disease, characterised by progressive growth and development of renal cysts that destroy the functioning parenchyma. It affects approximately one out of 1,000 people and is responsible for 7-10% of the cases of chronic kidney disease (CKD) that require substitutive renal treatment.1
ADPKD is caused by mutations in the PKD1 gene, located in chromosome 16 (16p 13.3), responsible for 85-90% of the cases, in the PKD2 gene, located in chromosome 4 (4q21-23), responsible for 10-15% of the cases, and possibly, in a third gene, PKD3, that has not yet been identified.2 There are, in addition, patients with mutations in both genes, trans-heterozygous mutations in PKD1 and PKD2, with more dismal clinical evolution than those with mutation in only one of these genes. Both forms of ADPKD have similar pathogenesis and similar clinical features, but in patients with mutation in PKD2 clinical manifestations appear later and progression toward terminal nephropathy occurs 10 years later than in patients with mutation in PKD1; besides, these patients have longer life expectancy (69.1 against 53).
The proteins coded by PKD1 and PKD2 genes are called polycystin-1 and polycystin-2, respectively.3,4 They are transmembrane proteins that act as mechanosensors and signal transducers, and regulate proliferation, adhesion, migration, differentiation, and cellular maturation. The mutations of the genes mentioned above produce defective proteins, which cause uncontrollable tissue growth and liquid accumulation inside the cysts.
ADPKD is a multisystemic disease, with renal and extrarenal manifestations derived from renal cyst formation, and that in many cases it also causes cysts in the liver and pancreas.5
ADPKD diagnosis is usually done by ultrasound.6-9 Genetic diagnosis can be used as a complementary test in patients with family history for an early diagnosis of the disease, even in absence of symptoms, usually during the first or second decade of life.10-12 Genetic tests are performed to reach a definite diagnosis of ADPKD in patients with familial antecedents, and in patients with no antecedents of the disease but clinically indicative of ADPKD to confirm or rule out that the cysts are related to ADPKD. Other advantages linked to the genetic study would be to offer advise of genetic nature with certainty regarding reproductive ages, which consists in informing the patient of the existence or not of the disease, how it is inherited and the risks associated with transmitting it to descendants,1,13,14 and to consider organ donation from relatives to affected patients. Molecular diagnosis may not predict the onset, severity, type of symptoms or progression of the disease; however, an early diagnosis of ADPKD would result in a better prognosis, allowing a more rigorous clinical follow-up.15
The genetic diagnosis may be performed by directly searching for the mutation or indirectly, by linkage analysis.16-19 Mutational analysis poses difficulties due to the large size and complexity of the PKD1 gene, and to the large number of mutations and polymorphisms described in such gene, which makes difficult to distinguish pathogenic changes from neutral changes. A diagnostic option for these patients would be, therefore, genetic linkage analysis, which requires many affected family members.
Given the difficulty for a genetic diagnosis in patients with ADPKD and mutation in the PKD1 gene and in spite of its higher prevalence, in our study, we considered carrying out a mutational analysis of the PKD2 gene in unrelated live patients clinically and radiologically diagnosed with ADPKD, with aims at comparing benefits of genetic against radiological studies, perform an early diagnosis for ADPKD in descendants of affected patients, and finally, try to set a phenotype-genotype correlation in patients with PKD2 mutation.
MATERIAL AND METHODS
Population of study
We have carried out the genetic study over 18 out of 48 patients with ADPKD that required medical assistance at the Nephrology Department of the University Hospital of Salamanca during the period between 1994-2005 and followup until April 2008; one member of each family knew for certain of the hereditary character of this disease. Samples from live family members were taken from those patients in whom mutation of the PKD2 gene was observed. The patients had been diagnosed with ADPKD according to clinical and radiological criteria. The samples were taken after informed consent.
Genetic Study
10ml of peripheral blood were withdrawn by venipuncture to obtain the DNA for the molecular study.
Exons of the PKD2 gene were then amplified by PCR. Exon 1 was not amplified due to difficulties found in PCR amplification because of abundance of guanine and cytosine.
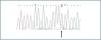
Once amplified, the fragments underwent a CSGE heteroduplex analysis to determine the presence of any mutation in the gene coding region (figure 1).
All PCR products where variants were detected were sequenced.
Nnpredict bioinformatic application was used to check whether the mutations found were responsible or not for structural changes within the protein, and, Esefinder to check whether the mutations were or not in exonic regions that regulate RNA processing.
Clinical follow-up
Main clinical aspects of patients and relatives with ADPKD where the PKD2 gene mutation was observed were analysed. Clinical histories provided information on all these data. The main parameters analysed were age of disease onset, aspects related to initial clinical manifestations of the disease, to clinical manifestations occurring during evolution of the disease, toextra-renal manifestations, and to survival and morbidity and mortality.
The qualitative variables were expressed as percentages and numerical variables as mean standard deviation (X ± SD).
RESULTS
Genetic analysis
A genetic study was performed in search of mutations in PKD2, in 18 patients, 9 women and 9 men. Each one of these patients belonged to families where there was certainty as to the hereditary character of ADPKD.
The only finding was encountered in a man, in whom a substitution of the adenosine nucleotide for cytokine was detected in position 2398 (c.2398 A>C) (figure 2), which implied a change in the methionine amino acid (Met) for leucine (Leu) in position 800 (p.800 Met>Leu).
The study of mutated polycystin-2 by computing methods did not show any structural changes in the protein.
The mutation did not lie in any exonic region regulating RNA processing (figure 3).
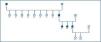
Once the genealogical tree had been made and the blood samples from live family members taken, the same finding was confirmed in his 2 sisters, who had the same clinical condition, but not in his daughters, who were healthy (figure 4).
Clinical analysis of patients with mutation in PKD2
Once the genealogical tree of patients with mutation in the PKD2 gene was made, the hereditary character of the disease was confirmed (figure 4). Transmission had taken place through the father and there was record of the disease in the paternal grandfather (figure 4), both deceased.
Of the 3 live patients affected, 2 were women and 1 was a man. Diagnosis mean age was 33 ± 3.79 years. (table 1). Renal failure, present in 100% of mutation carriers was the disease presenting symptom in two of them; other symptoms at the moment of diagnosis were abdominal pain (2 patients), macroscopic haematuria (1 patient) and urinary tract infections (2 patients). As disease progressed the patients developed renal lithiasis (1 patient), hyperuricaemia (2 patients), haematuria in 100% of the patients with a mean age occurrence of 51.5 ± 0.71 years, HT in 100%, and terminal stage chronic kidney disease (CKD) in 100%, with mean age occurrence of 35.25 ± 24.35 years and time elapsed from diagnosis of CKD of 4.8 ± 6.91 years. The only extra-renal manifestation was 2 of the 3 patients affected who presented with liver cysts detected by ultrasound at the moment of diagnosis (table 1).
Once the clinical and genetic studies were performed, ADPKD was ruled out in the descendants.
DISCUSSION
Genetic diagnosis
An early diagnosis is possible by using imaging diagnosis techniques in patients with familial antecedents of ADPKD, even in absence of symptoms, usually during the first or second decade of life.6-9,16-18 However, no familial antecedents were found in around 40% of the patients, which suggests either a high rate of de novo mutation, or the existence of modifying genes that affect the disease expression,20 which would make an early diagnosis difficult for these patients. Genetic diagnosis is an increasingly used alternative.
Both forms of ADPKD have similar pathogenesis and similar clinic; although in ADPKD with mutation in PKD2 clinical manifestations and progression to terminal nephropathy occur later and patients have longer life expectation,21 there are no phenotypic features that allow us to differentiate at the moment of diagnosis between each other. Thus the importance of resorting to the genetic study as a tool for early diagnosis, especially in families with mutation in the PKD2 gene, where genetic analysis is more sensitive than ultrasound study, mostly over the first decades of life, which permits diagnosing the condition before renal cysts and clinical symptoms occur.16
In our study, the prevalence of patients with ADPKD and diagnosed with mutations in PKD2 was lower than that described (5.56 against 10-15%),22,23 which suggests a higher prevalence of patients with mutation in PKD1, which in turn would agree with clinical evolution toward an early staging of terminal CKD in the patients under study, or in a biased selection of patients included in the genetic study. Given that ADPKD with PKD2 mutation is clinically detectable later and we only analysed those patients for whom there was certainty as to the hereditary character of the disease, it may have occurred that we did not genetically analysed patients with ADPKD and parents with mutation in the PKD2 gene not diagnosed with ADPKD due to presenting with mild clinical manifestations of the disease or because they died early without having been diagnosed with the disease.
Given the high prevalence of CKD and terminal stage CKD secondary to ADPKD in our study, early diagnosis of ADPKD would result in a better prognosis in relation with a more rigorous follow-up. In this respect, the molecular diagnosis offers the possibility of an early intervention regarding follow-up and treatment of HT, infections or lithiasis and, consequently, it delays CKD occurrence and reduce incidence of patients requiring substitutive renal treatment and co-morbidity that may result in renal failure and ultimately mortality. An interesting point in relation with early diagnosis is to carry a follow-up on these patients to know whether there is a tendency in some families or individuals to manifest early, and whether these initial symptoms have an adverse effect on the evolution of the disease or not.
Analysis of mutation and family segregation
In our study, after the genetic analysis was performed, a family with mutation in exon 13 in the PKD2 gene was diagnosed, and a change of the adenosine nucleotide to cytosine was observed in position 2398 (c.2398 A>C), which implied a change in the methionine amino acid (Met) to leucine (Leu) in position 800 (p.800 Met>Leu).
The meaning of such mutation has not been determined and although it has already been described by other groups that observed that this mutation did not segregate with the disease,10,20 we have, on the contrary, found out that all those members clinically and by imaging techniques diagnosed with ADPKD did present with such mutation. Consequently, we disagree with the studies just mentioned and confirm through ours that such mutation segregates with the disease.
Polycystin-1 and polycystin-2 interact via C-terminal end,24,25 mediated by the coiled-coil domain of polycystin-2 located, according to various publications, in exon 12 and 13 of polycystin-226 or in the polycystin-2 region between codon 872 and the carboxy terminal extreme (exons 14 and 15).27 It seems that patients with mutation at the extreme 3¿ present with less renal complications (HT, haematuria, renal lithiasis, urinary infections, etc.) as the mutated polycystin can preserve the function.28 These data, referred to in the literature, lead us to consider that the mutation found in our population has scant clinical significance. The structural study of the protein by bio-computing methods confirmed absence of structural significance, probably because such protein affects an important domain in the protein structure. The analysis of the exonic region where the mutation occurred determined that such region did not regulate RNA processing.
In our patients the mutation had clinical significance and all the patients with mutation in exon 13 behaved clinically as patients with mutation in the PKD1 gene and evolved rapidly toward renal failure. This leads us to consider that the members of the affected family may present, in addition, a mutation in the PKD1 gene (trans-heterozygous mutation), which would account for the most severe clinical course of the disease, not justified by an isolated mutation neither in the PKD1 gene nor in the PKD2 gene.27 Absence of either clinical or genetic manifestations in the descendants does not wholly rule out the disease, as imaging technique diagnosis at the third decade of life does not offer concluding results and the analysis of the PKD1 gene in further research would be needed to rule out the disease in the descendants, who were healthy at the moment of this study.
We were unable to confirm any clinical-genetic correlation in our work, a non-conclusive result given the small number of patients affected by the mutation in the PKD2 gene, and the evolutionary pattern between the affected members varied greatly. This heterogeneity may be attributed to the existence of modified genes that increase the severity of the defect.20
Given the high prevalence of patients with ADPKD with late diagnosis staging advanced CKD and terminal CKD, it is necessary to include the early genetic diagnosis in the protocols as a tool to reduce complications associated with the disease.
In short, mutations in the PKD2 gene are a minor cause of ADPKD in our environment. For this reason we consider of utmost importance to analyse the PKD1 gene in order to be able to perform a genetic diagnosis in these patients.

Figure 1.

Figure 2.

Figure 3.

Figure 4.

Table 1. Clinical characteristics of live members of the family with ADPKD and mutation in PKD2



