


Presentamos 3 pacientes con sospecha clínica de poliquistosis renal autosómica dominante (PQRAD)1, pero con la colaboración del grupo UMERH-RM se consiguió el diagnóstico correcto de síndrome orofaciodigital tipo I (SOFDI).
Caso 1. Mujer de 25 años con antecedentes personales de déficit cognitivo e intervenciones quirúrgicas de prognatismo, maloclusión abierta anterior con extracción de 2 caninos supernumerarios y otras piezas dentarias, fibromas y frenillos en la lengua. Se deriva a nefrología por hipertensión, filtrado glomerular (FG) de 61ml/min y ecografía renal: riñón derecho de 11cm e izquierdo de 12,4cm, múltiples quistes corticomedulares e hígado con pequeños quistes, compatible con PQRAD. En ecografía realizada 5 años antes, no había quistes renales y los riñones medían 10,3 y 10,8cm.
A la exploración física: acné miliar, fisuras palpebrales inclinadas, hipoplasia de alas nasales, apiñamiento dental, prognatismo, clino-braquidactilia en manos y pies y hallux valgus.
En la resonancia magnética (RM) cerebral se identificó agenesia de cuerpo calloso e hipoplasia del vermis cerebeloso.
En el estudio genético, hallazgo de variante patogénica de novo por deleción en exón 12: c.1193_1196delAATC (p.Q398LfsX1) en gen OFD1, dando lugar a proteína truncada, descrita previamente asociada al SOFDI.
Caso 2. Mujer de 32 años con enfermedad renal crónica y FG de 47ml/min, en contexto de abuso de antiinflamatorios no esteroideos por hernia discal. Intervenida en la infancia de frenillos gingivales accesorios y extracción de piezas dentarias supernumerarias por maloclusión.
Tras derivarse a nefrología en 2015, se objetivan riñones poliquísticos en la ecografía: riñón derecho de 13cm e izquierdo de 13,4cm, sin alteraciones en hígado, bazo o páncreas.
Antecedentes familiares: madre trasplantada renal por consumo crónico de antiinflamatorios no esteroideos por artritis reumatoide. El resto de familiares (padre, hermana, hijo e hija) con función renal normal. Todos se realizaron ecografía con riñones de tamaño normal y sin quistes.
A la exploración física: fisura del labio superior, micrognatia, paladar ojival y lengua lobulada y braqui-clinodactilia con marcada diferencia de tamaño entre ambas manos.
En el seguimiento apareció hipertensión y rápido deterioro del FG: 37ml/min a los 6 meses y FG: 30ml/min al año. En nueva ecografía, aumento de tamaño renal (derecho de 14,7 e izquierdo de 15,1cm). Diagnosticada clínicamente como PQRAD de novo, rápida progresadora y candidata a tolvaptán. Se solicitó RM cerebral y facial, por vértigos y cefaleas, sin encontrar anomalías. Actualmente tiene 37 años y FG:14ml/min.
El estudio de secuenciación masiva por Next Generation Sequencing (NGS) de enfermedades quísticas renales identificó variante patogénica en exón 2 del gen OFD1: c.71dup, (p.Try24*), no descrita previamente, dando lugar a una proteína truncada; cambiando el diagnóstico de PQRAD por el de SOFDI. El estudio familiar confirmó la misma variante en su hija, descartándola en el resto de familiares.
Caso 3. Mujer de 12 años, hija del caso 2. Sin antecedentes de interés, salvo la extracción de un canino supernumerario a los 6 años. En la exploración física: frenillo accesorio y manos con braquidactilia y clinodactilia (como su madre, aunque en menor grado de afectación) (fig. 1). En la ecografía los riñones fueron de tamaño normal y sin quistes.
 y caso 3 (hija). En la madre (izquierda) observamos braquidactilia y clinodactilia, mientras que en la hija (derecha) están presentes en menor medida.")
El SOFDI (OMIN #311200; ORPHA 2750) es una ciliopatía2, con una prevalencia: 1/50.000-1/250.000 en recién nacidos vivos.
En 1998 De Conciliis3 identificó como causa al gen OFD1 en el cromosoma X, que codifica proteína de 1.011 aminoácidos, expresada en centrosoma y cuerpo basal de los cilios primarios4. En el SOFDI se altera el proceso de embriogénesis causando dismorfias y quistes renales. Existen hasta 18 tipos de síndrome orofaciodigital, pero el tipo i es el más frecuente5.
Hasta el 75% de variantes patogénicas del gen OFD1 son esporádicas o de novo. En los casos familiares, el patrón de herencia es dominante ligado al cromosoma X, siendo letal en hombres afectados durante la gestación6, por lo que se trasmitiría de madres a hijas.
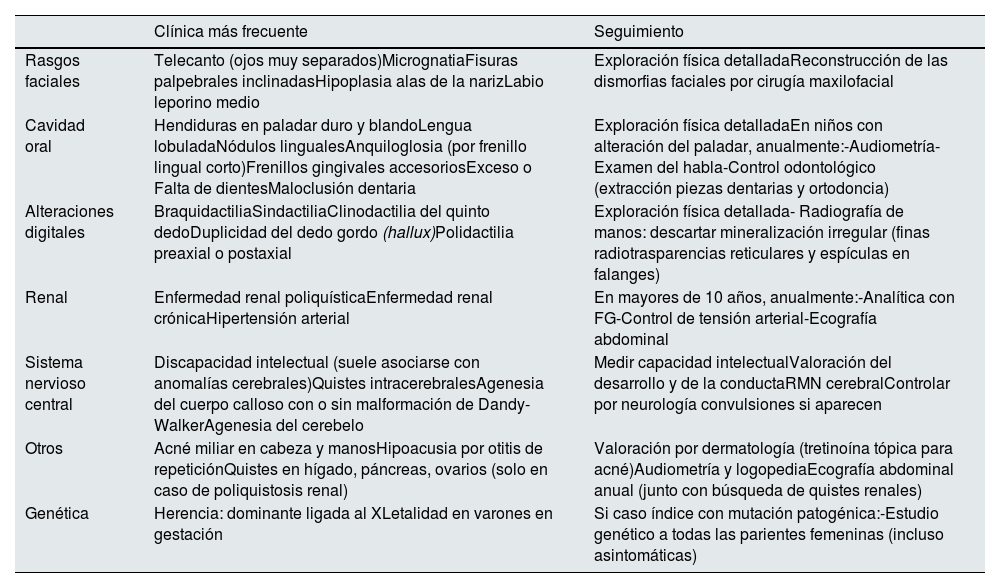
El diagnóstico clínico de sospecha del SOFDI se establece tras el nacimiento por las dismorfias orofaciales y digitales; o en adultos por asociarse riñones poliquísticos (tabla 1)7. Se recomienda control anual de tensión arterial, función renal y ecografía abdominal porque en el 50%, o hasta en la mayoría de mujeres según otros autores6, aparecen riñones poliquísticos8 y puede ser la única manifestación.
Principales características clínicas y seguimiento
| Clínica más frecuente | Seguimiento | |
|---|---|---|
| Rasgos faciales | Telecanto (ojos muy separados)MicrognatiaFisuras palpebrales inclinadasHipoplasia alas de la narizLabio leporino medio | Exploración física detalladaReconstrucción de las dismorfias faciales por cirugía maxilofacial |
| Cavidad oral | Hendiduras en paladar duro y blandoLengua lobuladaNódulos lingualesAnquiloglosia (por frenillo lingual corto)Frenillos gingivales accesoriosExceso o Falta de dientesMaloclusión dentaria | Exploración física detalladaEn niños con alteración del paladar, anualmente:-Audiometría-Examen del habla-Control odontológico (extracción piezas dentarias y ortodoncia) |
| Alteraciones digitales | BraquidactiliaSindactiliaClinodactilia del quinto dedoDuplicidad del dedo gordo (hallux)Polidactilia preaxial o postaxial | Exploración física detallada- Radiografía de manos: descartar mineralización irregular (finas radiotrasparencias reticulares y espículas en falanges) |
| Renal | Enfermedad renal poliquísticaEnfermedad renal crónicaHipertensión arterial | En mayores de 10 años, anualmente:-Analítica con FG-Control de tensión arterial-Ecografía abdominal |
| Sistema nervioso central | Discapacidad intelectual (suele asociarse con anomalías cerebrales)Quistes intracerebralesAgenesia del cuerpo calloso con o sin malformación de Dandy-WalkerAgenesia del cerebelo | Medir capacidad intelectualValoración del desarrollo y de la conductaRMN cerebralControlar por neurología convulsiones si aparecen |
| Otros | Acné miliar en cabeza y manosHipoacusia por otitis de repeticiónQuistes en hígado, páncreas, ovarios (solo en caso de poliquistosis renal) | Valoración por dermatología (tretinoína tópica para acné)Audiometría y logopediaEcografía abdominal anual (junto con búsqueda de quistes renales) |
| Genética | Herencia: dominante ligada al XLetalidad en varones en gestación | Si caso índice con mutación patogénica:-Estudio genético a todas las parientes femeninas (incluso asintomáticas) |
Debido a su gran variabilidad fenotípica, se precisa confirmación genética. Los paneles por NGS de enfermedad renal quística, que en el 80% identifican la causa molecular, son la prueba de elección9.
En conclusión, todas las mujeres con SOFDI deben ser seguidas anualmente por nefrología, porque pueden desarrollar riñones poliquísticos y enfermedad renal crónica progresiva, que condiciona su pronóstico10. El SOFDI nos plantea el diagnóstico diferencial con la PQRAD, siendo clave la búsqueda de dismorfias orofaciales y digitales en la exploración física.
Conflicto de interesesTodos los autores declaran que no ha habido conflicto de intereses.



