


Las enfermedades renales hereditarias (ERH) son una causa frecuente de enfermedad renal crónica, habiéndose incrementado su diagnóstico desde la introducción de la secuenciación masiva (NGS). En 2018 se fundó la Unidad multidisciplinar de Enfermedades Renales Hereditarias de la Región de Murcia basándose en el estudio genético de las ERH mediante panel de genes. El objetivo de este estudio es analizar los resultados obtenidos en los primeros tres años de funcionamiento, así como analizar los factores clínicos que se asocian a la obtención de un diagnóstico genético final.
Materiales y métodosSe incluyeron los pacientes estudiados mediante panel de genes de ERH y se compararon las características entre los que obtuvieron un diagnóstico genético final y los que no.
ResultadosSe estudiaron un total de 360 pacientes, detectándose variantes genéticas en 164 pacientes (45,6%) no relacionados familiarmente. Cuarenta y cinco de estas variantes eran de significado clínico incierto precisando estudio de cosegregación familiar, facilitado por la unidad multidisciplinar. Globalmente, considerando los resultados obtenidos con el panel de NGS realizado en el CBGC y los estudios genómicos ampliados, se consiguió un rendimiento diagnóstico final de ERH del 33,3% (120/360), contando hallazgos incidentales, del 35,6% (128/360).
Se estudiaron 223 pacientes con sospecha de síndrome de Alport, confirmándose el diagnóstico en un 28,5% (gen más frecuente COL4A4), los cuales eran con más frecuencia mujeres, y con clara historia familiar compatible. También tenían con más frecuencia microhematuria, aunque 5 pacientes sin microhematuria confirmaron diagnóstico. No hubo diferencias en la edad, proteinuria, función renal, hipoacusia o alteraciones oftalmológicas. El hallazgo más frecuente en la biopsia renal fue la proliferación mesangial. Calculamos que 39 pacientes evitaron la realización de biopsia renal.
Se estudiaron también 101 pacientes por sospecha de PQR, un 49,5% tuvieron un resultado genético concluyente (gen más frecuente PKD1), con mayor frecuencia mujeres, con tamaños renales mayores (aunque 9 pacientes con tamaño renal normal confirmaron diagnóstico). De nuevo la característica más predictiva de resultado genético fue la historia familiar.
ConclusionesLa implementación de un panel de NGS para ERH, junto con el abordaje multidisciplinar de los casos, ha mejorado el rendimiento diagnóstico de las ERH. En nuestra muestra, hemos confirmado el síndrome de Alport autosómico dominante como el de mayor incidencia. Las exploraciones oftalmológicas y auditivas no contribuyeron al diagnóstico. Hemos visto un descenso importante en la indicación de biopsias renales gracias al diagnóstico molecular. El abordaje multidisciplinar, con la participación activa de nefrólogos, pediatras, genetistas clínicos y moleculares, con insistencia en el adecuado fenotipado del paciente y revisión de su historia familiar, ofrece una mejor interpretación de las variantes genéticas, permitiendo la reclasificación del diagnóstico de algunas nefropatías no filiadas como ERH, mejorando así su manejo y consejo genético.
Hereditary kidney diseases (HKD) are a frequent cause of chronic kidney disease, and their diagnosis has increased since the introduction of next generation sequencing (NGS). In 2018, the Multidisciplinary Unit for Hereditary Kidney Diseases of the Region of Murcia (UMERH-RM) was founded based on the genetic study of HKD. The objective of this study is to analyze the results obtained in the first 3 years of operation, and to analyze the clinical factors associated to a final genetic diagnosis.
Materials and methodsAll the patients studied with the HKD gene panel were included. The characteristics between those who obtained a final genetic diagnosis and those who did not were compared.
ResultsA total of 360 patients were studied, detecting genetic variants in 164 not related patients (45.6%). 45 of these were variants of uncertain significance requiring a family co-segregation study, which was facilitated by the multidisciplinary unit. Overall, considering the results obtained with the NGS panel and the extended genomic studies, a final diagnostic yield of HRD of 33.3% (120/360) was achieved, and including incidental findings 35.6% (128/360).
Two hundred and twenty-three patients with suspected Alport syndrome were studied. Diagnosis was confirmed in 28.5% (COL4A4 most frequent gene), more frequently women with an obvious compatible family history. They also had frequently microhematuria, although 5 patients without microhematuria confirmed the diagnosis. There were no differences in age, proteinuria, renal function, hearing loss, or ophthalmologic abnormalities. The most frequent finding in the renal biopsy was mesangial proliferation. We estimate that 39 patients avoided renal biopsy.
A total of 101 patients with suspected PKD were also studied, 49.5% had a conclusive genetic result (most frequent gene PKD1), more frequently women, with larger kidney sizes (although 9 patients with normal kidney size confirmed diagnosis). Again, the most predictive characteristic of genetic outcome was family history.
ConclusionsThe implementation of an NGS panel for HKD, together with the multidisciplinary approach to cases, has improved the diagnostic performance of HKD. In our sample, autosomal dominant Alport syndrome is of highest incidence. Ophthalmological and auditory examinations did not contribute to the diagnosis. We have seen a significant decrease in the indication of renal biopsies thanks to molecular diagnosis. The multidisciplinary approach, with the active participation of nephrologists, paediatricians, clinical and molecular geneticists, with insistence on adequate patient phenotyping and review of their family history, offers a better interpretation of genetic variants, allowing reclassification of the diagnosis of some nephropathies, thus improving their management and genetic advice.
Las enfermedades renales hereditarias (ERH) son una causa frecuente de enfermedad renal crónica (ERC), constituyendo el 9% de los casos de enfermedad renal crónica terminal (ERCT)1, y siendo a menudo infradiagnosticadas como nefropatías no filiadas2. Las ERH más frecuentes son la poliquistosis renal autosómica dominante (PQRAD) y el síndrome de Alport (SA). Su diagnóstico ha sido tradicionalmente clínico, principalmente mediante los hallazgos en pruebas de imagen en el caso de la PQRAD, y los de la biopsia renal en el caso del SA, junto con los antecedentes familiares. El inicio del estudio genético de estas enfermedades supuso un apoyo para el diagnóstico, consiguiéndose la detección de casos infradiagnosticados. En los últimos años, la introducción de la secuenciación masiva (next generation sequencing [NGS]) en los estudios moleculares, ha permitido una mejora en el rendimiento diagnóstico de las ERH.
La PQRAD es la ERH más frecuente, causante de hasta el 6-7% de ERCT1. Su diagnóstico por imagen es sencillo en gran parte de los casos. Sin embargo, existen casos de diagnóstico dudoso, casos esporádicos y casos de enfermedad atípica, para los que el estudio molecular constituye la base diagnóstica de la enfermedad3. Un diagnóstico precoz permite iniciar medidas que han demostrado ralentizar su progresión, como el control de la presión arterial (PA) o el aumento de ingesta hídrica, y detectar aquellos pacientes con rápida progresión, susceptibles de iniciar tratamiento farmacológico4.
El SA es una ERH causante del 1-2% de los casos de inicio de terapia renal sustitutiva (TRS)5,6. Se desarrolla por defectos en el correcto ensamblaje de las cadenas α3, α4 y α5 del colágeno tipo IV en la membrana basal glomerular, codificados en los genes COL4A3, COL4A4 y COL4A57,8. El SA clásico consiste en hematuria seguida de proteinuria y de enfermedad renal progresiva, asociada a hipoacusia neurosensorial y, en algunos pacientes, alteraciones oculares como lenticono, retinopatía o leiomatosis9. Su forma sindrómica se encuentra mayoritariamente en varones con síndrome de Alport ligado al X (SALX, debido a mutaciones en el gen COL4A5) y en los casos de SA autosómico recesivo (SAAR, producido por variantes bialélicas en los genes COL4A3 o COL4A4). Las mujeres con variantes patogénicas en COL4A5 presentan un fenotipo variable debido al fenómeno de inactivación del cromosoma X10. Por su parte, el espectro clínico informado de los pacientes con variantes patogénicas en heterozigosis en los genes COL4A3 y COL4A4 es muy amplio, desde microhematuria aislada hasta enfermedad renal proteinúrica progresiva11,12, sugiriéndose la reclasificación como SA autosómico dominante (SAAD) a cualquier paciente con variantes patogénicas heterocigóticas en genes COL4A3 y COL4A4, independientemente de la clínica en el momento del diagnóstico13, facilitado por el aumento importante de los casos diagnosticados genéticamente en los últimos años mediante NGS14. El diagnóstico de esta enfermedad permite el tratamiento temprano con bloqueantes del sistema renina-angiotensina-aldosterona, asociado con mejoría pronóstica en niños incluidos en el ensayo EARLY-PROTECT15 y en estudios observacionales en adultos16. Actualmente, existen además otros fármacos en fase de investigación12.
En el año 2018, se inició en la Región de Murcia (1,5 millones de habitantes) el diagnóstico de las principales ERH mediante un panel de genes de NGS. Estos estudios moleculares se centralizaron en el Centro de Bioquímica y Genética Clínica (CBGC) del Hospital Virgen de la Arrixaca (Murcia). Para mejorar la calidad asistencial del paciente, se creó la Unidad Multidisciplinar de Enfermedades Renales Hereditarias de la Región de Murcia (UMERH-RM), con participación de nefrólogos, pediatras, genetistas clínicos y genetistas moleculares. Este grupo realiza reuniones periódicas en las que se discuten casos clínicos y se proponen proyectos y objetivos comunes. A través de este grupo se lleva a cabo la caracterización genética de los pacientes con sospecha de ERH, gracias a la realización de un panel de NGS que incluye genes asociados a ERH y, en aquellos casos que se consideran pertinentes por el grupo, se lleva a cabo la ampliación del estudio mediante exoma clínico o mediante un panel para ERH que incluye mayor número de genes implicados en enfermedades renales. A través de este grupo, se deciden además los estudios familiares pertinentes, los de casos complejos y se valora el consejo genético de casos especiales. Todo ello ha llevado a un incremento en la demanda de estudios genéticos por parte de los clínicos, con un aumento importante de la detección de casos en nuestra región y una disminución en la demora diagnóstica.
Sin embargo, los estudios genéticos conllevan la detección de variantes de significado incierto o dan lugar en ocasiones a resultados dudosos, lo que aumenta la necesidad de un estudio familiar clínico y genético completo. También, pueden conllevar la detección de hallazgos incidentales que, en ocasiones, requieren de una reevaluación clínica. Por todo ello, resulta de vital importancia analizar los resultados obtenidos desde la instauración del grupo multidisciplinar y cohesionarlos con la clínica de los pacientes y de sus familiares.
Así, el objetivo principal de este estudio es analizar los resultados obtenidos mediante técnicas genómicas a lo largo de los tres años de trabajo de la UMERH-RM. Como objetivos secundarios planteamos analizar los factores clínicos que se asocian a la obtención de un diagnóstico genético final, revisar los hallazgos incidentales encontrados y estimar la reducción conseguida en pruebas invasivas gracias a la obtención de un diagnóstico molecular concluyente.
Material y métodosEl diseño del estudio es observacional retrospectivo y multicéntrico. Se han recopilado datos demográficos, clínicos, analíticos y genéticos.
Criterios de inclusiónSe incluyeron los pacientes procedentes de los hospitales de la Región de Murcia que cuentan con servicio de Nefrología, a los que se les realizó un estudio genético desde el inicio de la implementación de un panel de NGS de ERH en el CBGC (septiembre de 2018) hasta diciembre de 2021.
Criterios de exclusiónFamiliares de casos índices diagnosticados, en los que solo se realizó secuenciación Sanger de la variante familiar.
Pacientes que firmaron su negativa al uso de sus datos de forma anónima en estudios de investigación, dentro del consentimiento informado de realización de la prueba genética (se adjunta).
Variables clínicasSe recogieron los datos clínicos de edad en el momento de diagnóstico genético, antecedentes familiares (contexto familiar general, número de familiares con clínica renal, número de familiares que han llegado a terapia renal sustitutiva [TRS]), alteraciones oftalmológicas o auditivas (hipoacusia global e hipoacusia con caída en tonos agudos), antecedentes de realización de biopsia renal y sus resultados, tamaño renal en ecografía y presencia de quistes renales.
Variables analíticasSe recogieron variables analíticas en el momento del estudio genético: presencia de hematuria y cuantificación en hematíes por campo (H/C), creatinina (Cr) sérica, filtrado glomerular estimado (FGe) según la fórmula Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI)17 o la fórmula Schwartz bedside en edad pediátrica18, cociente de albúmina/creatinina (CAC).
Análisis genéticoA lo largo de este estudio se emplearon dos paneles de captura de diseño Agilent, que incluían sondas específicas para genes relacionados con las ERH más frecuentes. La preparación de librerías se realizó mediante los kits comerciales SureSelect QXT® o SureSelect XT HS® (Agilent Technologies) y la posterior secuenciación en un equipo MiSeq (Illumina). El primer panel, utilizado hasta marzo de 2021, incluía los genes de ERH: COL4A1, COL4A3, COL4A4, COL4A5, PKD1, PKD2, PKHD1, HNF1B. El segundo panel, usado desde marzo de 2021, amplió el número de genes asociados a ERH, añadiendo al anterior los genes MYH9,DNAJB11, GANAB, DZIP1L, UMOD, REN, ACE, AGT, AGTR1, NPHP1 y GLA. Estos paneles incluían además otros genes asociados a otras enfermedades genéticas no relacionados con ERH.
El tipo de panel empleado fue conocido y evaluado por la unidad multidisciplinar a la hora de esclarecer el diagnóstico. Para el filtrado, anotación y análisis de variantes se empleó el software bioinformático Alissa Interpret (Agilent Technologies). La interpretación de las variantes identificadas se realizó de acuerdo con las Directrices del Colegio Americano de Genética Médica y Genómica19 y se informaron solamente las variantes patogénicas, probablemente patogénicas y de significado clínico incierto. La confirmación y segregación familiar de dichas variantes se realizó mediante secuenciación Sanger (Abi Prism 3130, Sequencing Analysis Software 6, Applied Biosystems). Ante un resultado negativo con alta sospecha de ERH, se realizó MLPA (para la identificación de posibles variantes en el número de copia), panel de NGS ampliado20 o exoma clínico. Estos dos últimos tipos de estudios genéticos se realizaron en un laboratorio externo.
Análisis estadísticoLas variables cualitativas se describieron mediante frecuencia y porcentajes, y las cuantitativas mediante media y desviación estándar. La comparación de características clínicas entre los grupos de pacientes se realizó mediante el test chi-cuadrado para las variables cualitativas. En el caso de las variables cuantitativas se aplicó el test t de Student para aquellas con distribución normal o test no paramétricos para datos distribución no normal. La correlación de Spearman se utilizó para analizar la correlación entre medidas paramétricas y no paramétricas. Para el análisis estadístico se utilizó el programa SPSS® versión 22. Se consideró estadísticamente significativo un valor p<0,05.
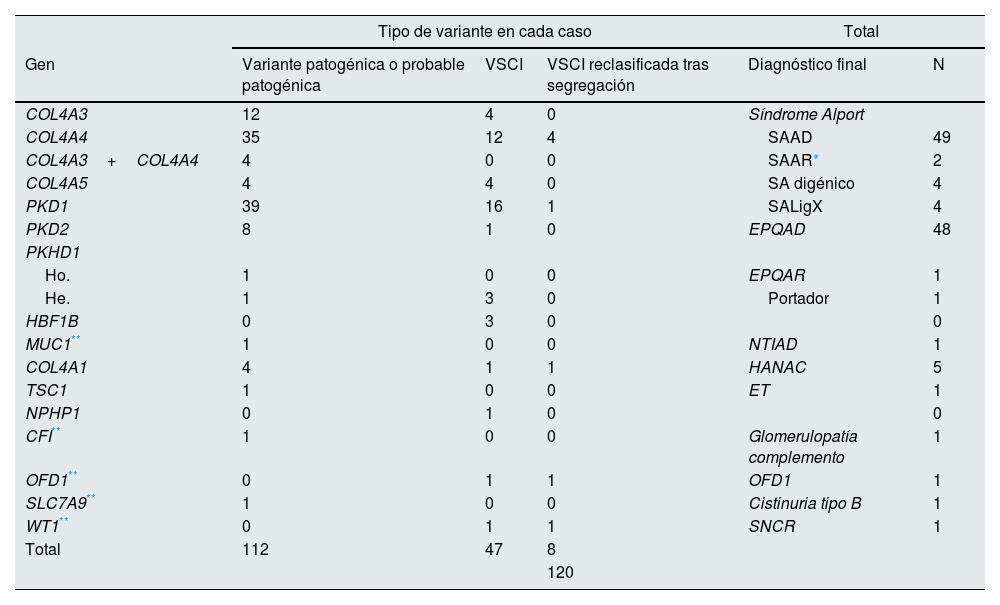
ResultadosEstudio genético de los pacientesDurante 3 años y 3 meses de actividad, se han estudiado un total de 360 pacientes con el panel de NGS. Se han detectado variantes genéticas en 164 pacientes (45,6%) no relacionados familiarmente (tabla 1 y fig. 1).
Casos con variantes genéticas en NGS
| Tipo de variante en cada caso | Total | ||||
|---|---|---|---|---|---|
| Gen | Variante patogénica o probable patogénica | VSCI | VSCI reclasificada tras segregación | Diagnóstico final | N |
| COL4A3 | 12 | 4 | 0 | Síndrome Alport | |
| COL4A4 | 35 | 12 | 4 | SAAD | 49 |
| COL4A3+COL4A4 | 4 | 0 | 0 | SAAR* | 2 |
| COL4A5 | 4 | 4 | 0 | SA digénico | 4 |
| PKD1 | 39 | 16 | 1 | SALigX | 4 |
| PKD2 | 8 | 1 | 0 | EPQAD | 48 |
| PKHD1 | |||||
| Ho. | 1 | 0 | 0 | EPQAR | 1 |
| He. | 1 | 3 | 0 | Portador | 1 |
| HBF1B | 0 | 3 | 0 | 0 | |
| MUC1** | 1 | 0 | 0 | NTIAD | 1 |
| COL4A1 | 4 | 1 | 1 | HANAC | 5 |
| TSC1 | 1 | 0 | 0 | ET | 1 |
| NPHP1 | 0 | 1 | 0 | 0 | |
| CFI** | 1 | 0 | 0 | Glomerulopatía complemento | 1 |
| OFD1** | 0 | 1 | 1 | OFD1 | 1 |
| SLC7A9** | 1 | 0 | 0 | Cistinuria tipo B | 1 |
| WT1** | 0 | 1 | 1 | SNCR | 1 |
| Total | 112 | 47 | 8 | ||
| 120 | |||||
EPQAD: enfermedad poliquística autosómica dominante; EPQAR: enfermedad poliquística autosómica recesiva; ET: esclerosis tuberosa; HANAC: síndrome de hematuria familiar –tortuosidad arteriolar retiniana– contracturas; He.: heterozigosis; Ho.: homocigosis; SA: síndrome de Alport; SAAD: síndrome de Alport autosómico dominante; SAAR: síndrome de Alport autosómico recesivo; SALigX: síndrome de Alport ligado al X; SN: síndrome nefrótico; SNCR: síndrome nefrótico córtico-resistente. VSCI: variante de significado clínico incierto.

En 108 pacientes (30,0%) se identificaron variantes patogénicas o probable patogénicas en genes asociados a ERH. De ellos, 55 variantes se encontraban en genes asociados a SA (32 pacientes con variantes monoalélicas en COL4A4, 10 monoalélicas en COL4A3, 4 monoalélicas en COL4A5, 4 digénicas en COL4A3/4, 3 con dos variantes en COL4A4, 2 con dos variantes en COL4A3), 47 en genes asociados a EPQAD (39 PKD1, 8 PKD2), 4 en el gen COL4A1 (asociado a síndrome de hematuria familiar autosómica dominante con tortuosidad arteriolar retiniana y –contracturas– HANAC), 1 en homocigosis en el gen PKHD1, asociado a poliquistosis autosómica recesiva - PQAR), 1 en TSC1 (asociado a esclerosis tuberosa (tabla 1 y fig. 1).
Se hallaron en total 45 variantes de significado clínico incierto (VSCI) que precisaron estudio de cosegregación de la variante familiar. En 11 de ellas este estudio no fue posible, dada la escasa informatividad de algunas familias. En 6 se observó que la variante cosegregaba con los signos clínicos renales, por lo que a dichas variantes se les atribuyó un efecto causal de la enfermedad, siendo 4 de ellas en el gen COL4A4 y 1 en PKD1. En otras 6 de las VSCI, el análisis familiar no mostró cosegregación, por lo que estas variantes no se consideraron informativas para la familia, o se reclasificaron como probablemente benignas (1 en COL4A1, 2 en COL4A5, 2 en HNF1B, 1 en PKD1). Las otras 22 VSCI se encuentran pendientes del estudio familiar en el momento de la escritura de este manuscrito.
Por otro lado, se encontraron otras variantes patogénicas o probablemente patogénicas relacionadas con ERH, pero que no correspondían al fenotipo de sospecha, o para las que el paciente era portador heterocigoto para variantes en genes con herencia recesiva: 2 variantes probablemente patogénicas en COL4A1 en dos pacientes con poliquistosis (con variantes también en PKD1), 1 portador de variante patogénica en gen PKHD1, 1 portador de variante patogénica en SLC12A3 (síndrome de Gitelman AR), 1 variante probablemente patogénica en COL4A4 (sin fenotipo familiar de SAAD, precisando estudio familiar), 1 variante probable patogénica en PKD1 en paciente joven sin fenotipo de poliquistosis (lo que obliga a seguimiento y estudio familiar). Se identificaron también VSCI en genes de ERH no relacionados con el fenotipo de sospecha, por lo que, tras el reanálisis del caso clínico no se realizó estudio familiar (41 en PKD1, 11 en PKHD1 en heterocigosis, 4 en PKD2, 3 en COL4A1, 3 en COL4A3, 2 en COL4A4, 2 en HNF1B, 2 en EYA-1, 1 en COL4A5, 1 en SALL1, 1 en GANAB, 1 en MYH9, 1 en heterocigosis en NPHP1, 1 en UMOD, 1 en heterocigosis en ACE). No se encontraros variantes en los genes DNAJB11 ni ALG9.
En aquellos pacientes con panel negativo, pero con sospecha fundada de ERH, se solicitó el panel ampliado de ERH, encontrándose: una variante patogénica en el gen MUC1 (asociado a nefritis intersticial autosómica dominante-NTIAD), una variante probablemente patogénica en gen CFI (asociado a glomerulopatía del complemento), una variante probablemente patogénica en gen SLC7A9 (asociado a cistinuria tipo B) y una VSCI en WT1 (asociado a síndrome nefrótico genético resistente a corticoides, disgenesia gonadal y nefroblastoma).
Debido a que el panel de NGS para ERH contiene también otros genes no relacionados con enfermedad renal y a la ampliación de estudio mediante exoma de algunos pacientes, se pudieron identificar variantes patogénicas incidentales en los siguientes genes: JAG1 (síndrome de Alagille), PTPN11 (síndrome de Noonan), UROD (porfiria cutánea tarda AD), UROS (portador de porfiria eritropoyética congénita AR), BTD (portador heterocigoto de la deficiencia de biotinidasa), COL11A2 (portador de sordera neurosensorial no sindrómica), COL9A2 (displasia epifisaria múltiple por anomalía del colágeno 9 AD), COL21A2 (portador de hiperplasia suprarrenal congénita forma no clásica).
Globalmente, considerando los resultados obtenidos con el panel de NGS realizado en el CBGC y los estudios genómicos ampliados, se consiguió un rendimiento diagnóstico final de ERH del 33,3% (120/360), contando hallazgos incidentales, del 35,6% (128/360).
Estudio de los factores clínicos que se asocian a un resultado genético positivoSe compararon las características clínicas de los pacientes con resultado genético positivo frente a las observadas en los pacientes en los que no se obtuvo resultado.
Pacientes con sospecha inicial de SASe estudiaron un total de 223 pacientes con sospecha de enfermedad relacionada con el colágeno IV. Para el análisis comparativo, se excluyeron los pacientes pendientes de estudio familiar o con resultado final de otra ERH, quedando finalmente 207 pacientes no relacionados. De ellos, 59 (28,5%) tuvieron un resultado genético final positivo con variantes en genes asociados a dicha enfermedad (ampliable hasta 69 –33,3%–, de confirmarse la cosegregación de las VSCI en el estudio familiar). Los pacientes con resultado genético positivo eran en mayor frecuencia mujeres (69,1%), tenían una edad media al diagnóstico de 49,7±14,4 años (sin diferencias significativas con el grupo con resultado negativo). Los pacientes con SA confirmado tenían en mayor proporción microhematuria comparado con los de estudio negativo (91,1% vs. 78,4%, p=0,015). No hubo diferencias en la presencia de proteinuria (64% vs. 58,3%, p=0,0717), CAC medio (533±511mg/g vs. 519±498mg/g, p=0,906) y FGe medio al diagnóstico (71±33ml/min/1,73m2 vs. 81±21ml/min/1,73m2, p=0,463). No hubo tampoco diferencias significativas entre los dos grupos en la existencia de hipoacusia (27,9% vs. 24,5%, p=820), o hipoacusia con caída típica en sonidos agudos (20,6% vs. 14,4%, p=0,129) (teniendo en cuenta que tenían audiometría 104 pacientes), ni en alteraciones oftalmológicas (5,9% vs. 2,2%, p=0,392) (tenían exploración oftalmológica 86 pacientes). No hubo tampoco diferencias en presencia de quistes renales (33,8% vs. 23,2%, p=0,104) ni tampoco comparando entre sí los pacientes con proteinuria y sin proteinuria (0=0,600). Las principales diferencias entre grupos se encontraron en el contexto familiar: el 86,2% de los pacientes con resultado positivo tenían antecedentes familiares de ER, frente a un 44,60% en los pacientes sin resultado final (p=0,001). La media del número de familiares con clínica renal fue de 2,5±1,6 en el primer grupo y 1,6±1,6 en el segundo (p=<0,005), y la media de familiares en TRS fue de 0,7±0,6 y 0,4±0,3, respectivamente (p=0,06). Cinco pacientes sin microhematuria tuvieron diagnóstico genético de SA, todos ellos presentaban albuminuria y, al menos, un familiar de primer grado en TRS.
Se analizaron un total de 28 pacientes con biopsia renal realizada previamente al estudio genético. Dos de ellos contaban con análisis de microscopía electrónica, ambos con membrana basal adelgazada, y en ambos se consiguió un diagnóstico genético de SAAD. Del resto, otros 7 pacientes fueron confirmados genéticamente de SAAD y entre sus biopsias renales mostraban: 1 con glomérulo aparentemente normal, 1 glomeruloesclerosis focal y segmentaria (GEFS), y 5 con proliferación mesangial (dos de los cuales tenían depósitos de IgA y por tanto estaban diagnosticados de nefropatía mesangial IgA, otro tenía depósitos de IgM y otros dos no tenían depósitos). El resto de la muestra no tenía biopsia renal previa. Observando las indicaciones generales de biopsia renal en el resto de la muestra global, calculamos que en 39 pacientes se pudo evitar una biopsia renal gracias a la obtención de un diagnóstico genético previo.
Pacientes con sospecha inicial de PQRSe solicitó estudio genético a un total de 101 pacientes por sospecha de PQR. Los criterios para solicitar estudio genético en la EPQAD fueron la duda diagnóstica en los fenotipos no clásicos o atípicos de EPQAD, los casos esporádicos sin antecedentes familiares, los casos en los que tener un diagnóstico de certeza era necesario pero no era definitivo por la prueba de imagen en ese momento (como en un posible donante de vivo joven) o cuando se requería para un adecuado consejo genético reproductivo. De los 101 pacientes, un 49,5% tuvieron un resultado genético concluyente: 40 con variantes en PKD1, 8 en PKD2, 1 en PKHD1 en homocigosis y 1 en COL4A4 (aunque la sospecha inicial fue de poliquistosis). De los pacientes con PQRAD con diagnóstico genético, 64% eran mujeres. El FGe en el momento del diagnóstico fue de 76±34ml/min/1,73m2, el CAC 78±22mg/g, ambos sin diferencias respecto al grupo con diagnóstico genético negativo. Todos los pacientes tenían quistes renales. Entre los pacientes con genética positiva, el tamaño renal era grande (60%) o muy grande (20%), con diferencia respecto a los pacientes sin diagnóstico genético, en los que un 72% tenían tamaño normal (p<0,001). De los 32 pacientes con tamaño renal normal estudiados, 9 confirmaron diagnóstico de PQRAD, teniendo 5 de ellos un contexto familiar claro, pero 4 de ellos sin antecedentes familiares previos, y teniendo todos variantes en gen PKD1, excepto un paciente con una variante en PKD2, en el que sí existía historia familiar de ER. No se detectaron diferencias significativas en la edad al diagnóstico entre el grupo de pacientes con tamaño renal normal y con tamaño renal grande o muy grande (41±21 años vs. 42±16 años, p=0,808). De forma general, el contexto familiar predijo de nuevo un resultado genético positivo: el 80% de los pacientes con confirmación genética tenían un contexto familiar claro, mientras que solo el 44% de los pacientes sin confirmación lo tenían (p<0,001).
DiscusiónLa instauración en nuestra región del diagnóstico de las principales ERH mediante herramientas genómicas y la creación de un grupo pluridisciplinar de nefropatías hereditarias ha aumentado de forma considerable el diagnóstico de pacientes con nefropatía no filiada. Desde su inicio en 2018 hasta final de 2021 se han llevado a cabo 360 estudios, con un rendimiento del 32,5%, aumentando este rendimiento al 35,6% cuando se incluyen los pacientes con estudio ampliado o con diagnósticos incidentales. Estimamos que hemos conseguido un 5-7% de mayor rendimiento gracias al análisis detallado de las VSCI y el enfoque multidisciplinar del Grupo UMERH.
La disponibilidad de dicho panel de genes en el laboratorio del CBGC del Hospital Virgen de la Arrixaca (Murcia) ha permitido una mayor accesibilidad al diagnóstico genético. El panel de genes está diseñado para detectar las ERH más frecuentes, lo que ha permitido diagnosticar un elevado número de pacientes (tabla 1). Esto ha reducido los costes del diagnóstico y ha agilizado su resultado en este grupo de pacientes, al no hacer necesario el envío de muestras a un centro de referencia (esta opción se reserva para aquellos casos con panel negativo y alta sospecha de ERH).
Sin embargo, esta mayor accesibilidad puede llevar a un aumento exponencial de solicitudes de estudio genético, lo que contribuye al hallazgo de un número creciente de variantes de significado clínico incierto de difícil análisis, ya sea por la dificultad de encontrar familias informativas, por la reducida penetrancia de determinadas variantes y/o por su baja o nula relación con el fenotipo del paciente. También conlleva la identificación de un mayor número de hallazgos incidentales, en ocasiones variantes patogénicas en homocigosis de enfermedad AR. Todo ello implica un incremento de estudios familiares y la necesidad de reevaluación de casos. Para hacer frente a este volumen, nuestra UMERH-RM realiza reuniones mensuales o bimensuales en las que se exponen y discuten distintos casos y familias. La colaboración de nefrólogos/as, pediatras, genetistas clínicos y genetistas moleculares ha permitido un mejor análisis de los casos, siendo una de las principales conclusiones la elevada importancia de una adecuada caracterización fenotípica de los pacientes para el correcto análisis de los resultados genéticos personales y familiares.
En nuestra población, confirmamos una mayor incidencia de SAAD en los últimos años, con mayor frecuencia en mujeres. El SALX ha sido tradicionalmente considerado el más frecuente (hasta un 80% de los casos), aunque es aceptado que la prevalencia de las mutaciones en genes COL4A3/4 es desconocida21. La mayor detección de estos casos contribuye a un manejo temprano de la patología y de su posible evolución a enfermedad renal proteinúrica progresiva12,22.
Por otra parte, se debe tratar de configurar un algoritmo para depurar el exceso de detección de variantes sin significado clínico. En nuestra muestra, las variables mayoritariamente asociadas a un resultado genético positivo han sido el contexto familiar y la presencia de microhematuria. Todos los pacientes que sin microhematuria confirmaron SA, presentaban albuminuria, descenso del FGe y, al menos, un familiar de primer grado en TRS. Por ello, parece recomendable incluir entre las indicaciones de estudio genético con sospecha de SA, la ERC proteinúrica de causa no filiada con algún familiar con ERC (especialmente ERCT), aunque no esté presente la microhematuria. Esto se corrobora en otros estudios en los que, al analizar la genética de pacientes con ERC no filiada, se encontró un elevado porcentaje de mutaciones en los genes COL4A3/4/52,23. No hemos encontrado una asociación significativa con las alteraciones auditivas (ni siquiera en la hipoacusia con caída en agudos típica de la enfermedad) u oculares, seguramente asociado a una muy menor prevalencia de estas alteraciones en el SAAD13, por lo que no parece recomendable basar la solicitud de un estudio genético en la presencia o ausencia de estas alteraciones, ni realizar estudios indiscriminados sin confirmación de la enfermedad.
Cabe destacar el rendimiento de un 32% para el diagnóstico de SA entre pacientes con biopsia renal previa practicada, con histologías de GEFS y de GN proliferativa mesangial con y sin depósitos inmunes, como se describe en otros artículos publicados24, aunque queda por dilucidar si la asociación con la nefropatía mesangial IgA es únicamente casual. Por otro lado, en este estudio hemos estimado que en 39 pacientes con diagnóstico genético concluyente se ha podido evitar la realización de una biopsia renal. Por tanto, podría ser de utilidad evaluar la posibilidad de un estudio genético antes de indicar la biopsia renal, para lo que resulta de suma importancia la realización de un árbol familiar detallado en la evaluación nefrológica inicial, así como reevaluar algunos casos previamente biopsiados, sobre todo en los casos de hallazgos glomerulares compatibles, resistentes a tratamiento y/o con antecedentes familiares.
Por otra parte, entre los pacientes con sospecha de poliquistosis, el rendimiento del estudio genético fue del 49,5%. De nuevo, la variable de contexto familiar, así como el fenotipo ecográfico de poliquistosis, se asociaron a un resultado genético positivo. Cabe destacar un rendimiento del 28,1% entre los pacientes sin fenotipo claro de PQRAD, con múltiples quistes, pero tamaño renal normal, casi la mitad de los cuales no tenían un contexto familiar claro. La gran mayoría de ellos presentaban variantes en PKD1 y no había diferencias en la edad respecto del grupo con fenotipo claro. Por tanto, parece razonable indicar el estudio genético con sospecha de PQRAD en este grupo de pacientes, como primera posibilidad, antes de buscar otros genes menos frecuentes.
ConclusionesLa implementación de un panel de NGS para el estudio de las ERH más frecuentes en la Región de Murcia, junto con el abordaje multidisciplinar de los casos, ha conseguido un rendimiento diagnóstico global del 35,6%. Sin embargo, debido a que las técnicas genómicas se asocian a la detección de un mayor número de variantes de valor clínico desconocido y de hallazgos incidentales, se deberían afinar los criterios para la indicación del estudio genético. En todos los casos prima la existencia de contexto familiar claro, aunque hay casos confirmados de SA sin contexto familiar en pacientes con enfermedad renal proteinúrica, incluso sin microhematuria; y casos confirmados de PQRAD en pacientes con tamaño renal normal de riñones multiquísticos. Las exploraciones oftalmológicas y auditivas no contribuyeron al diagnóstico. De forma adicional, hemos visto un descenso importante en la indicación de biopsias renales gracias al diagnóstico molecular.
En nuestra muestra, el gen mayoritariamente asociado a SA ha sido el COL4A4, seguido de COL4A3, lo que hace el SAAD el de mayor incidencia. En el caso de EPQAD ha sido PKD1, seguido de PKD2, como se ha descrito en otras poblaciones.
El abordaje multidisciplinar, con la participación activa de nefrólogos, pediatras, genetistas clínicos y moleculares, con insistencia en el adecuado fenotipado del paciente y revisión de su historia familiar, ofrece una mejor interpretación de las variantes genéticas, permitiendo la reclasificación del diagnóstico de algunas nefropatías no filiadas como ERH, mejorando así su manejo y consejo genético.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



