



Hereditary kidney diseases (HKD) are a frequent cause of chronic kidney disease, and their diagnosis has increased since the introduction of next generation sequencing (NGS). In 2018, the Multidisciplinary Unit for Hereditary Kidney Diseases of the Region of Murcia (UMERH-RM) was founded based on the genetic study of HKD. The objective of this study is to analyze the results obtained in the first 3 years of operation, and to analyze the clinical factors associated to a final genetic diagnosis.
Materials and methodsAll the patients studied with the HKD gene panel were included. The characteristics between those who obtained a final genetic diagnosis and those who did not were compared.
ResultsA total of 360 patients were studied, detecting genetic variants in 164 not related patients (45.6%). 45 of these were variants of uncertain significance requiring a family co-segregation study, which was facilitated by the multidisciplinary unit. Overall, considering the results obtained with the NGS panel and the extended genomic studies, a final diagnostic yield of HRD of 33.3% (120/360) was achieved, and including incidental findings 35.6% (128/360).
Two hundred and twenty-three patients with suspected Alport syndrome were studied. Diagnosis was confirmed in 28.5% (COL4A4 most frequent gene), more frequently women with an obvious compatible family history. They also had frequently microhematuria, although 5 patients without microhematuria confirmed the diagnosis. There were no differences in age, proteinuria, renal function, hearing loss, or ophthalmologic abnormalities. The most frequent finding in the renal biopsy was mesangial proliferation. We estimate that 39 patients avoided renal biopsy.
A total of 101 patients with suspected PKD were also studied, 49.5% had a conclusive genetic result (most frequent gene PKD1), more frequently women, with larger kidney sizes (although 9 patients with normal kidney size confirmed diagnosis). Again, the most predictive characteristic of genetic outcome was family history.
ConclusionsThe implementation of an NGS panel for HKD, together with the multidisciplinary approach to cases, has improved the diagnostic performance of HKD. In our sample, autosomal dominant Alport syndrome is of highest incidence. Ophthalmological and auditory examinations did not contribute to the diagnosis. We have seen a significant decrease in the indication of renal biopsies thanks to molecular diagnosis. The multidisciplinary approach, with the active participation of nephrologists, paediatricians, clinical and molecular geneticists, with insistence on adequate patient phenotyping and review of their family history, offers a better interpretation of genetic variants, allowing reclassification of the diagnosis of some nephropathies, thus improving their management and genetic advice.
Las enfermedades renales hereditarias (ERH) son una causa frecuente de enfermedad renal crónica, habiéndose incrementado su diagnóstico desde la introducción de la secuenciación masiva (NGS). En 2018 se fundó la Unidad multidisciplinar de Enfermedades Renales Hereditarias de la Región de Murcia basándose en el estudio genético de las ERH mediante panel de genes. El objetivo de este estudio es analizar los resultados obtenidos en los primeros tres años de funcionamiento, así como analizar los factores clínicos que se asocian a la obtención de un diagnóstico genético final.
Materiales y métodosSe incluyeron los pacientes estudiados mediante panel de genes de ERH y se compararon las características entre los que obtuvieron un diagnóstico genético final y los que no.
ResultadosSe estudiaron un total de 360 pacientes, detectándose variantes genéticas en 164 pacientes (45,6%) no relacionados familiarmente. Cuarenta y cinco de estas variantes eran de significado clínico incierto precisando estudio de cosegregación familiar, facilitado por la unidad multidisciplinar. Globalmente, considerando los resultados obtenidos con el panel de NGS realizado en el CBGC y los estudios genómicos ampliados, se consiguió un rendimiento diagnóstico final de ERH del 33,3% (120/360), contando hallazgos incidentales, del 35,6% (128/360).
Se estudiaron 223 pacientes con sospecha de síndrome de Alport, confirmándose el diagnóstico en un 28,5% (gen más frecuente COL4A4), los cuales eran con más frecuencia mujeres, y con clara historia familiar compatible. También tenían con más frecuencia microhematuria, aunque 5 pacientes sin microhematuria confirmaron diagnóstico. No hubo diferencias en la edad, proteinuria, función renal, hipoacusia o alteraciones oftalmológicas. El hallazgo más frecuente en la biopsia renal fue la proliferación mesangial. Calculamos que 39 pacientes evitaron la realización de biopsia renal.
Se estudiaron también 101 pacientes por sospecha de PQR, un 49,5% tuvieron un resultado genético concluyente (gen más frecuente PKD1), con mayor frecuencia mujeres, con tamaños renales mayores (aunque 9 pacientes con tamaño renal normal confirmaron diagnóstico). De nuevo la característica más predictiva de resultado genético fue la historia familiar.
ConclusionesLa implementación de un panel de NGS para ERH, junto con el abordaje multidisciplinar de los casos, ha mejorado el rendimiento diagnóstico de las ERH. En nuestra muestra, hemos confirmado el síndrome de Alport autosómico dominante como el de mayor incidencia. Las exploraciones oftalmológicas y auditivas no contribuyeron al diagnóstico. Hemos visto un descenso importante en la indicación de biopsias renales gracias al diagnóstico molecular. El abordaje multidisciplinar, con la participación activa de nefrólogos, pediatras, genetistas clínicos y moleculares, con insistencia en el adecuado fenotipado del paciente y revisión de su historia familiar, ofrece una mejor interpretación de las variantes genéticas, permitiendo la reclasificación del diagnóstico de algunas nefropatías no filiadas como ERH, mejorando así su manejo y consejo genético.
Inherited renal diseases (HKD) are a common cause of chronic kidney disease (CKD), accounting for 9% of cases of end-stage renal disease (ESRD),1 and are often underdiagnosed as non-inherited nephropathies.2 The most frequent HKD are autosomal dominant polycystic dominant renal disease (ADPKD) and Alport syndrome (AS). Their diagnosis has traditionally been clinical, mainly through imaging test findings in the case of ADPKD, and renal biopsy findings in the case of AS, together with the family history. The beginning of the genetic study of these diseases was a support for diagnosis, achieving the detection of underdiagnosed cases. In recent years, the introduction of massive sequencing (next generation sequencing [NGS]) in molecular studies has allowed an improvement in the diagnostic yield of HKD.
ADPKD is the most frequent HKD, causing up to 6%–7% of ESRD.1 Its diagnosis by imaging is simple in most cases. However, there are cases of doubtful diagnosis, sporadic cases and cases of atypical disease, for which the molecular study constitutes the basis for diagnosis of the disease.3 Early diagnosis makes it possible to initiate measures that have been shown to slow progression, such as blood pressure (BP) control or increased water intake, and to detect those patients with rapid progression, who are susceptible to pharmacological treatment.4
The AS is an HKD that leads to 1%–2% of cases that initate renal replacement therapy (RRT).5,6 It develops due to defects in the correct assembly of the chains α 3, α 4 and α 5 of type IV collagen in the glomerular basement membrane, encoded in the COL4A3, COL4A4 and COL4A5 genes.7,8 Classic AS consists of hematuria followed by proteinuria and progressive renal disease, associated with sensorineural hearing loss and, in some patients, ocular alterations such as lenticonus, retinopathy or leiomatosis.9 Its syndromic form is mostly found in males with X-linked Alport syndrome (XLAS, due to mutations in the COL4A5 gene) and in cases of autosomal recessive AS (ARAS, caused by biallelic variants in the COL4A3 or COL4A4 genes). Women with pathogenic variants in COL4A5 have a variable phenotype due to the X chromosome inactivation phenomenon.10 For their part, the reported clinical spectrum of patients with pathogenic variants in heterozygosis in the COL4A3 and COL4A4 genes is very broad, ranging from isolated microhematuria to progressive proteinuric renal disease,11,12 suggesting the reclassification as autosomal dominant AS (ADAS) to any patient with pathogenic heterozygous variants in COL4A3 and COL4A4 genes, regardless of the clinical presentation at the time of diagnosis,13 facilitated by the significant increase in the number of cases diagnosed genetically in recent years by NGS.14 The diagnosis of this disease allows early treatment with renin–angiotensin–aldosterone system blockers, which is associated with prognostic improvement in children included in the EARLY-PROTECT trial15 and in observational studies in adults.16 Other drugs are currently under investigation.12
In 2018, the diagnosis of the main HKD using an NGS gene panel was initiated in the Region of Murcia (1.5 million inhabitants). These molecular studies were centralized at the Center for Biochemistry and Clinical Genetics (CBCG) of the Hospital Virgen de la Arrixaca (Murcia). To improve the quality of patient care, it was created the Multidisciplinary Unit for Hereditary Kidney Diseases of the Region of Murcia (MUHKD-RM) with the participation of nephrologists, pediatricians, clinical geneticists and molecular geneticists. This group holds periodic meetings in which clinical cases are discussed and common projects and objectives are proposed. Through this group, the genetic characterization of patients with suspected HKD is carried out, thanks to the performance of an NGS panel that includes genes associated with HKD and, in those cases considered relevant by the group, the study is extended by means of a clinical exome or by means of a panel for HKD that includes a greater number of genes involved in renal diseases. This group also decides on the pertinent family studies, complex cases and assesses the genetic counseling of special cases. All this has led to an increase in the demand for genetic studies by clinicians, with a significant increase in the detection of cases in our region and a decrease in the diagnostic delay.
However, genetic studies lead to the detection of variants of uncertain significance or sometimes give rise to equivocal results, increasing the need for a complete clinical and genetic family study. They can also lead to the detection of incidental findings that sometimes require clinical reevaluation. For all these reasons, it is of vital importance to analyze the results obtained since the establishment of the multidisciplinary group and to bring them into line with the clinical findings of the patients and their families.
Thus, the main objective of this study is to analyze the results obtained by genomic techniques during the three years of work at the MUHKD-RM.
Material and methodsThe present study is retrospective, observational and multicenter. Demographic, clinical, analytical and genetic data were collected.
Inclusion criteriaPatients were from Nephrology Services of hospitals in the Region of Murcia, who underwent a genetic study from the beginning of the implementation of a NGS panel of HKD at the CBCG (September 2018 until December 2021).
Exclusion criteriaRelatives of diagnosed index cases, in which only Sanger sequencing of the familial variant was performed.
Patients who signed their refusal to the use of their data anonymously in research studies, as part of the informed consent for genetic testing (attached).
Clinical variablesThere were collected clinical data of age at the time of genetic diagnosis, family history (general family context, number of family members with renal symptoms, number of family members who have undergone renal replacement therapy [RRT]), ophthalmologic or auditory alterations (global hearing loss and hearing loss with drop in acute tones), renal biopsy and its results, renal size by ultrasound and presence of renal cysts.
Analytical variablesAnalytical variables were collected at the time of the genetic study: the presence of hematuria and its quantification in red blood cells per field (H/C), serum creatinine (Cr), estimated glomerular filtration rate (eGFR) according to the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula17 or the Schwartz bedside formula in pediatric age,18 albumin/creatinine ratio (ACC).
Genetic analysisThroughout this study, two Agilent-designed capture panels were used, which included specific probes for genes related to the most frequent HKDs. The preparation of libraries was performed using commercial kits SureSelect QXT® or SureSelect XT HS® (Agilent Technologies) and subsequent sequencing on a MiSeq machine (Illumina). The first panel, used until March 2021, included the HKD genes COL4A1, COL4A3, COL4A4, COL4A4, COL4A5, PKD1, PKD2, PKHD1, HNF1B. The second panel, used since March 2021, expanded the number of HKD-associated genes, adding MYH9, DNAJB11, GANAB, DZIP1L, UMOD, REN, ACE, AGT, AGTR1, NPHP1 and GLA to the previous panel. These panels also included other genes associated with other genetic diseases unrelated to HKD.
The type of panel used was recognized and evaluated by the multidisciplinary unit to clarify the diagnosis. The Alissa Interpret bioinformatics software (Agilent Technologies) was used for filtering, annotation and analysis of variants. Interpretation of identified variants was performed according to the American College of Medical Genetics and Genomics Guidelines19 and only pathogenic, probably pathogenic and variants of uncertain clinical significance were reported. Confirmation and familial segregation of these variants was performed by Sanger sequencing (Abi Prism 3130, Sequencing Analysis Software 6, Applied Biosystems). In case of a negative result with high suspicion of HKD, it was performed a MLPA (multiplex ligation-dependent probe amplification) for the identification of possible copy number variants, and an extended NGS panel20 or clinical exome was also performed. These last two types of genetic studies were performed in an external laboratory.
Statistical analysisQualitative variables were described by frequency and percentages, and quantitative variables by mean and standard deviation. Comparison of clinical characteristics between patient groups was performed using the chi-square test for qualitative variables. In case of quantitative variables, Student’s t-test was used for those with normal distribution or nonparametric tests for non-normal distribution data. Spearman’s correlation was used to analyze the correlation between parametric and nonparametric measures. SPSS® version 22 was used for the statistical analysis. A p < 0.05 value was considered statistically significant.
ResultsGenetic study of patientsDuring 3 years and 3 months of activity, a total of 360 patients have been evaluated with the NGS panel. Genetic variants were detected in 164 (45.6%) unrelated patients (Table 1 and Fig. 1).
Cases with genetic variants on NGS.
| Gen | Type of variant in each case | Total | |||
|---|---|---|---|---|---|
| Pathogenic or probable pathogenic variant | VUS | VUS reclassified after spin-off | Final diagnosis | N | |
| COL4A3 | 12 | 4 | 0 | Alport syndrome | |
| COL4A4 | 35 | 12 | 4 | ADAS | 49 |
| COL4A3 + COL4A4 | 4 | 0 | 0 | ARASa | 2 |
| COL4A5 | 4 | 4 | 0 | AS digenic | 4 |
| PKD1 | 39 | 16 | 1 | XLAS | 4 |
| PKD2 | 8 | 1 | 0 | ADPKD | 48 |
| PKHD1 | |||||
| Ho. | 1 | 0 | 0 | ARPKD | 1 |
| He. | 1 | 3 | 0 | Carrier | 1 |
| HBF1B | 0 | 3 | 0 | 0 | |
| MUC1b | 1 | 0 | 0 | ADTKD | 1 |
| COL4A1 | 4 | 1 | 1 | HANAC | 5 |
| TSC1 | 1 | 0 | 0 | TE | 1 |
| NPHP1 | 0 | 1 | 0 | 0 | |
| IFCb | 1 | 0 | 0 | Complement glomerulopathy | 1 |
| OFD1b | 0 | 1 | 1 | OFD1 | 1 |
| SLC7A9b | 1 | 0 | 0 | Cystinuria type B | 1 |
| WT1b | 0 | 1 | 1 | CRNS | 1 |
| Total | 112 | 47 | 8 | ||
| 120 | |||||
ADPKD: autosomal dominant polycystic kidney disease; ARPKD: autosomal recessive polycystic kidney disease; ET: tuberous sclerosis; HANAC: familial hematuria syndrome – retinal arteriolar tortuosity – contractures; He.: heterozygosis; Ho.SA: Alport syndrome; ADAS: autosomal dominant Alport syndrome; ARAS: autosomal recessive Alport syndrome; XLAS: X-linked Alport syndrome; NS: nephrotic syndrome; CRNS: cortico-resistant nephrotic syndrome; OFD1: oral-facial-digital syndrome type 1. VUS: variant of uncertain clinical significance.
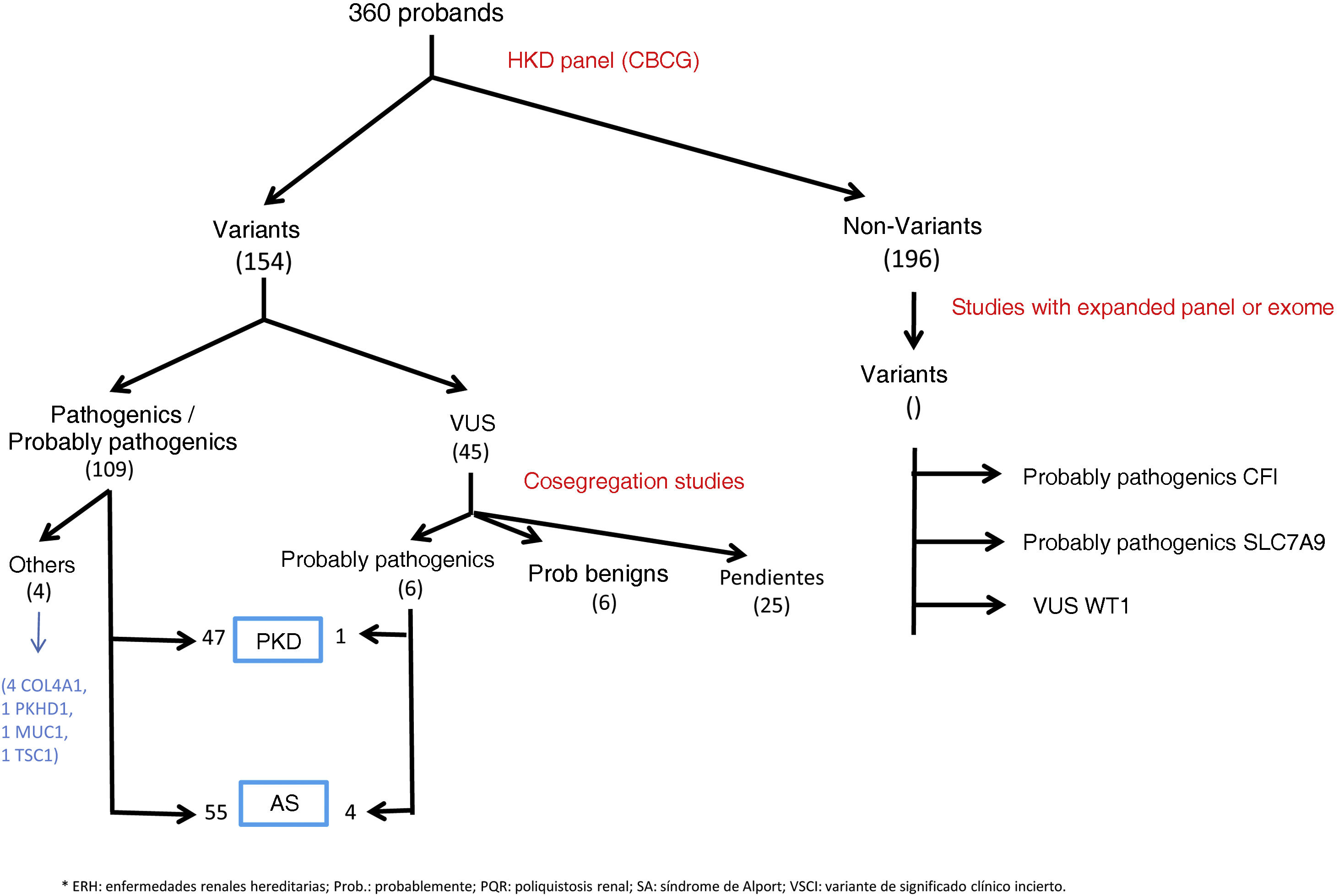
In 108 patients (30.0%) pathogenic or probable pathogenic variants were identified in genes associated with HKD. Of these, 55 variants were found in genes associated with AS (32 patients with monoallelic variants in COL4A4, 10 monoallelic in COL4A3, 4 monoallelic in COL4A5, 4 digenic in COL4A3/4, 3 with two variants in COL4A4, 2 with two variants in COL4A3), 47 in genes associated with ADPKD (39 PKD1, 8 PKD2), 4 in the COL4A1 gene (associated with autosomal dominant familial hematuria syndrome with retinal arteriolar tortuosity and –contractures– HANAC), 1 in homozygosity in the PKHD1 gene, associated with autosomal recessive polycystic kidney disease — ARPKD), 1 in TSC1 (associated with tuberous sclerosis (Table 1 and Fig. 1).
A total of 45 variants of uncertain clinical significance (VUS) were found that required a cosegregation study of the family variant. In 11 of them this study was not possible, given the poor informativity of some families. In 6 of them it was observed that the variant cosegregated with the renal clinical signs, so that a causal effect of the disease was attributed to these variants, 4 of them in the COL4A4 gene and 1 in PKD1. In 6 other VUS, the familial analysis did not show cosegregation, so these variants were not considered informative for the family, or were reclassified as probably benign (1 in COL4A1, 2 in COL4A5, 2 in HNF1B, 1 in PKD1). The other 22 VUSs are pending family study at the time of writing this manuscript.
In addition, there were found other pathogenic or probably pathogenic variants related to HKD, but they did not correspond to the suspected phenotype, or for which the patient was a heterozygous carrier for variants in genes with recessive inheritance. There were two probably pathogenic variants in COL4A1 in two patients with polycystic kidney disease (with variants also in PKD1), 1 carrier of pathogenic variant in PKHD1 gene, 1 carrier of pathogenic variant in SLC12A3 (Gitelman AR syndrome), 1 probably pathogenic variant in COL4A4 (without familial phenotype of SAAD, requiring family study), 1 probable pathogenic variant in PKD1 in a young patient without polycystic disease phenotype (which requires follow-up and family study). We also identified VUS in HKD genes not related to the suspected phenotype, so, after reanalysis of the clinical case, we did not perform a family study (41 in PKD1, 11 in PKHD1 in heterozygosis, 4 in PKD2, 3 in COL4A1, 3 in COL4A3, 2 in COL4A4, 2 in HNF1B, 2 in EYA-1, 1 in COL4A5, 1 in SALL1, 1 in GANAB, 1 in MYH9, 1 in heterozygosity in NPHP1, 1 in UMOD, 1 in heterozygosity in ACE). No variants were found in the DNAJB11 or ALG9 genes.
In those patients with a negative panel, but with a well-founded suspicion of HKD, the expanded HKD panel was requested and it was found: one pathogenic variant in the MUC1 gene (associated with autosomal dominant interstitial nephritis-ADTKD), one probably pathogenic variant in CFI gene (associated with complement glomerulopathy), one probably pathogenic variant in SLC7A9 gene (associated with cystinuria type B) and one VUS in WT1 (associated with genetic corticosteroid-resistant nephrotic syndrome, gonadal dysgenesis and nephroblastoma).
Due to the fact that the NGS panel for HKD also contains other genes not related to renal disease and to the extension of exome study of some patients, incidental pathogenic variants in the following genes wereb identified: JAG1 (Alagille syndrome), PTPN11 (Noonan syndrome), UROD (porphyria cutanea tarda AD), UROS (carrier of erythropoietic porphyria congenita AR), BTD (heterozygous carrier of biotinidase deficiency), COL11A2 (carrier of non-syndromic sensorineural deafness), COL9A2 (multiple epiphyseal dysplasia due to collagen 9 anomaly AD), COL21A2 (carrier of congenital adrenal hyperplasia non-classical form).
Overall, considering the results obtained with the NGS panel performed in the CBCG and the extended genomic studies, a final diagnostic yield of 33.3% (120/360) was achieved for HKD, and counting incidental findings it was 35.6% (128/360).
Study of the clinical factors associated with a positive genetic resultThe clinical characteristics of patients with a positive genetic result were compared with those observed in patients in whom no result was obtained.
Patients with initial suspicion of ASA total of 223 patients with suspected collagen IV-related disease were studied. For the comparative analysis, patients pending family study or with a final result of another HKD were excluded, finally leaving 207 unrelated patients. Of these, 59 (28.5%) had a positive final genetic result with variants in genes associated with this disease (potencially increased to 69 (33.3%), if the cosegregation of the VUS in the family study was confirmed). The patients with a positive genetic result were more frequently women (69.1%) and had a mean age at diagnosis of 49.7 ± 14.4 years (with no significant differences with the group with a negative result). Patients with confirmed AS had in higher proportion microhematuria compared to those with negative study (91.1% vs. 78.4%, p = 0.015). There were no differences in the presence of proteinuria (64% vs. 58.3%, p = 0.0717), mean CAC (533 ± 511 mg/g vs. 519 ± 498 mg/g, p = 0.906) and mean eGFR at diagnosis (71 ± 33 ml/min/1.73 m2 vs. 81 ± 21 ml/min/1.73 m2, p = 0.463). There was also no significant difference between the two groups in the existence of hypoacusis (27.9% vs. 24.5%, p = 820), or hypoacusis with typical drop in acute sounds (20.6% vs. 14.4%, p = 0.129) (taking into account that they had audiometry performed in104 patients), nor in ophthalmologic alterations (5.9% vs. 2.2%, p = 0.392) (86 patients had ophthalmologic examination). There were also no differences in the presence of renal cysts (33.8% vs. 23.2%, p = 0.104) or when patients with proteinuria and without proteinuria were compared (p = 0.600). The main differences between groups were found in the family context: 86.2% of patients with a positive result had a family history of kidney disease, compared to 44.60% in patients without a positive result (p = 0.001); the mean number of family members with clinical expression of renal diseases was 2.5 ± 1.6 in the first group and 1.6 ± 1.6 in the second group (p ≤ 0.005), and the mean number of relatives in RRT was 0.7 ± 0.6 and 0.4 ± 0.3, respectively (p = 0.06). Five patients without microhematuria had a genetic diagnosis of AS, all of them had albuminuria and at least one first-degree relative wa on RRT.
A total of 28 patients with renal biopsy performed prior to the genetic study were analyzed. Two of them had electron microscopy analysis, both with thinned basement membrane, and in both of them a genetic diagnosis of ADAS was obtained. Among the rest patients, 7 were genetically confirmed to have ADAS and their renal biopsies showed: 1 with apparently normal glomerulus, 1 with focal segmental glomerulosclerosis (FSGS), and 5 with mesangial proliferation (two of which had IgA deposits and therefore were diagnosed with IgA mesangial nephropathy, another had IgM deposits and another two had no deposits). The rest of the sample had no previous renal biopsy. Examining the general indications for renal biopsy in the rest of the overall sample, we calculated that in 39 patients a renal biopsy could be avoided by obtaining a previous genetic diagnosis.
Patients with initial suspicion of ADPKDA total of 101 patients with suspected ADPKD underwent genetic testing. The criteria for requesting genetic study in ADPKD were diagnostic uncertainties in non-classical or atypical phenotypes of ADPKD, sporadic cases with no family history, cases in which a diagnosis of certainty was necessary but was not definitive because of the imaging test at that time (as in a possible young living donor) or when it was required for adequate reproductive genetic counseling. Of the 101 patients, 49.5% had a conclusive genetic result: 40 with variants in PKD1, 8 in PKD2, 1 in PKHD1 in homozygosis and 1 in COL4A4 (although the initial suspicion was polycystic kidney disease). Of the ADPKD patients with a genetic diagnosis, 64% were women. The eGFR at diagnosis was 76 ± 34 ml/min/1.73 m2, the CAC 78 ± 22 mg/g, both without differences with respect to the group with negative genetic diagnosis. All patients had renal cysts. Among patients with positive genetics, renal size was large (60%) or very large (20%), with difference with respect to patients without genetic diagnosis, in which 72% had normal size (p < 0.001). Of the 32 patients studied with normal kidney size, 9 had confirmed the diagnosis of ADPKD, 5 of them had a clear family history, but 4 of them had no previous family history, and all had variants in the PKD1 gene, except for one patient with a variant in PKD2, in whom there was a family history of KD. No significant difference was detected in age at diagnosis between the group of patients with normal kidney size and with large or very large kidney size (41 ± 21 years vs. 42 ± 16 years, p = 0.808). Overall, family background again predicted a positive genetic result: 80% of patients with genetic confirmation had a clear family background, whereas only 44% of patients without confirmation did (p < 0.001).
DiscussionThe generation in our region of the diagnosis of the main HKDs using genomic tools and the creation of a multidisciplinary group of hereditary nephropathies has increased considerably the diagnosis of patients with unidentified nephropathy. Since its initiation in 2018 until the end of 2021, 360 studies have been performed, with a yield of 32.5%, increasing this yield to 35.6% when patients with extended study or incidental diagnoses are included. We estimate that we have achieved a 5%–7% higher yield due to the detailed analysis of VUS and the multidisciplinary approach of the UMHKD Group.
The availability of this gene panel in the CBCG laboratory of the Hospital Virgen de la Arrixaca (Murcia) has allowed greater accessibility to genetic diagnosis. The gene panel is designed to detect the most frequent HKD, which has made it possible to diagnose a large number of patients (Table 1). This has reduced the costs of diagnosis and has expedited the results in this group of patients, as it is not necessary to send samples to a reference center (this option is reserved for those cases with a negative panel and high suspicion of HKD).
However, this greater accessibility can lead to an exponential increase in requests for genetic studies, which contributes to the identification of an increasing number of variants of uncertain clinical significance that are difficult to analyze, either because of the difficulty to find informative families, the reduced penetrance of certain variants and/or their low or null relationship with the patient’s phenotype. It also leads to the identification of a greater number of incidental findings, sometimes pathogenic variants in homozygosis of AR disease. All this implies an increase in family studies and the need for reevaluation of cases. To cope with such a demand, our UMHKD-RM holds monthly or bimonthly meetings in which different cases and families are presented and discussed. The collaboration of nephrologists, pediatricians, clinical geneticists and molecular geneticists has allowed a better analysis of the cases, being one of the main conclusions the high importance of an adequate phenotypic characterization of the patients for the correct analysis of the personal and family genetic results.
In our population, we confirmed a higher incidence of ADAS in recent years, with a higher frequency in women. XLAS has traditionally been considered the most frequent (up to 80% of cases), although it is accepted that the prevalence of mutations in COL4A3/4 genes is unknown.21 The increased detection of these cases contributes to early management of the pathology and its possible evolution to progressive proteinuric renal disease.12,22
Additionally, we should try to configure an algorithm to purify the excessive detection of variants without clinical significance. In our sample, the variables most associated with a positive genetic result were the family background and the presence of microhematuria. All the patients who confirmed AS without microhematuria had albuminuria, decreased eGFR and at least one first-degree relative in RRT. Therefore, it seems advisable to include among the indications for genetic study with suspected AS, proteinuric CKD of non-filial cause with a family member with CKD (especially ES-CKD), even if microhematuria is not present. This is validated in other studies in which, when analyzing the genetics of patients with CKD of unknown etiology, a high percentage of mutations were found in the COL4A3/4/5 genes.2,23 We have not found a significant association with hearing disorders (not even in the hypoacusis with acute hearing loss typical of the disease) or ocular disorders, probably associated with a much lower prevalence of these disorders in the ADAS,13 so it does not seem advisable to base the request for a genetic study on the presence or absence of these disorders, nor to perform indiscriminate studies without confirmation of the disease.
Of note is the 32% yield for the diagnosis of AS among patients with previous renal biopsy performed, with histologies of FSGS and mesangial proliferative GN with and without immune deposits, as described in other published articles,24 although it remains to be elucidated whether the association with IgA mesangial nephropathy is only coincidental. On the other hand, in the present study we have estimated that in 39 patients with a conclusive genetic diagnosis it was possible to avoid performing a renal biopsy. Therefore, it could be useful to evaluate the possibility of a genetic study before indicating a renal biopsy, for which it is of utmost importance to perform a detailed family tree in the initial nephrological evaluation, as well as to reevaluate some cases previously biopsied, especially in cases of compatible glomerular findings, resistant to treatment and/or with family history.
Among patients with suspected polycystic disease, the yield of the genetic study was 49.5%. Again, the family background variable, as well as the ultrasound phenotype of polycystic disease, were associated with a positive genetic result. Of note was a 28.1% yield among patients with no clear ADPKD phenotype, multiple cysts, but normal renal size, almost half of whom had no clear familial background. The great majority of them had variants in PKD1 and there was no difference in age with respect to the group with clear phenotype. Therefore, it seems reasonable to indicate genetic study with suspicion of ADPKD in this group of patients, as a first possibility, before searching for other less frequent genes.
ConclusionsThe implementation of an NGS panel for the study of the most frequent HKD in the Region of Murcia, together with the multidisciplinary approach, has achieved an overall diagnostic yield of 35.6%. However, due to the fact that genomic techniques are associated with the detection of a greater number of variants of unknown clinical value and incidental findings, the criteria for the indication of the genetic study should be reconsidered. In all cases, the existence of a clear familial background is essential, although there are confirmed cases of AS without familial background in patients with proteinuric renal disease, even without microhematuria; and confirmed cases of ADPKD in patients with normal renal size of multicystic kidneys. Ophthalmologic and auditory examinations did not contribute to the diagnosis. Additionally, we have seen a significant decrease in the indication for renal biopsies due to molecular diagnosis.
In our sample, the gene most frequently associated with AS was COL4A4, followed by COL4A3, making ADAS the one with the highest incidence. In the case of ADPKD it was PKD1, followed by PKD2, as has been described in other populations.
The multidisciplinary approach, with the active participation of nephrologists, pediatricians, clinical and molecular geneticists, with insistence on adequate phenotyping of the patient and review of his family history, offers a better interpretation of genetic variants, allowing the reclassification of the diagnosis of some non-filial nephropathies as HKD, thus improving their management and genetic counseling.
Conflict of interestThe authors declare that they have no conflicts of interest.



