



Presentamos el caso de un paciente de 23 años, con 3 meses de evolución de cefalea, pérdida de la visión, hiporexia y orina espumosa. En la última semana: postración en cama, alteración del sensorio, oliguria y disnea. En la evaluación inicial se encontró en malas condiciones generales, somnoliento, desorientado, taquicárdico, hipertenso, taquipneico, con asterixis; aliento urémico e ingurgitación yugular; ruidos respiratorios con crépitos bibasales y edema de miembros inferiores. Pruebas de laboratorio al ingreso: azoados elevados, acidosis metabólica con anión GAP alto, anemia hemolítica microangiopática no inmune y trombocitopenia (tabla 1). Se solicitaron estudios complementarios, incluida actividad de ADAMTS13, y perfil inmunológico e infeccioso, que fueron negativos (tabla 1). Se inició tratamiento con hemodiálisis, labetalol parenteral, transfusión de glóbulos rojos y recambios plasmáticos. Una vez controlada la uremia y la crisis hipertensiva se realizó una evaluación oftalmológica que reportó pérdida marcada de la agudeza visual (OD: 20/400+1; OI: 20/150−1) y retinopatía hipertensiva. Al séptimo día se controló la actividad hemolítica, lo que permitió suspender los recambios plasmáticos; sin embargo, presentó recaída 48h después, razón por la cual se reiniciaron nuevamente y se obtuvo el diagnóstico definitivo de SHUa (fig. 1). Se inició tratamiento con eculizumab y se suspendieron los recambios plasmáticos, con adecuada evolución posterior, con recuperación de su visión (20/30 en ambos ojos), aunque continuó con requerimiento de diálisis. Se realizó estudio genético para detectar mutaciones del complemento asociadas con SHUa, el cual fue negativo para las mutaciones conocidas (tabla 1). En el seguimiento a 9 meses el paciente continuaba dependiente de diálisis, por lo cual se diagnosticó enfermedad renal crónica terminal (ERCT) secundaria a SHUa y se activó para trasplante renal.
Resultados de laboratorio
| Citoquímico de orina | Aspecto | pH | Densidad | Proteinuria | Eritrocitos | Leucocitos | Proteinuria |
| Turbio | 8 | 1,015 | >300mg/dl | >50CAP | 21-50CAP | 850mg/día | |
| Gases capilares | pH | HCO3 | PCO2 | PO2 | PAFI | Lactato | Exceso bases |
| 7,24 | 9,6mmol/l | 22mmHg | 121mmHg | 242 | 1,3mmol | −15,7mmol/l | |
| Perfil renal y metabólico | Creatinina | BUN | Ácido úrico | Sodio | Potasio | Cloro | Fósforo |
| 19,7mg/dl | 128mg/dl | 8,5mg/dl | 140mEq/l | 4,2mEq/l | 100mEq/l | 8mg/dl | |
| Calcio corregido | Calcio ionizado | Magnesio | Albúmina | Glucemia | Colesterol total | Colesterol-HDL | |
| 6,7mg/dl | 0,65mmol/l | 1,53mg/dl | 2,8g/dl | 92mg/dl | 108mg/dl | 31mg/dl | |
| Colesterol-LDL | Triglicéridos | Vitamina D | PTH | Vitamina B12 | Ferritina | Ácido fólico | |
| 63,6mg/dl | 61mg/dl | 19,3ng/ml | 751pg/ml | 286pg/ml | 551ng/ml | 7,7ng/ml | |
| Perfil hematológico | Hemoglobina | Hematocrito | Leucocitos | Neutrófilos | Linfocitos | Monocitos | Basófilos |
| 5,1g/dl | 14% | 6.200mm3 | 80,2% | 11,3% | 8,1% | 0,4% | |
| Eosinófilos | Plaquetas | Reticulocitos corregido | ESP | LDH | Haptoglobina | Actividad ADAMTS13 | |
| 0% | 82.000mm3 | 5,2% | Esquistocitos ++ | 547u/l | 10,6mg/dl | 74,5% | |
| TPT | Control TPT | TP | INR | Fibrinógeno | |||
| 30s | 26,1 | 11,3s | 1,04 | 435mg/dl | |||
| Perfil hepático | BT | BD | BI | AST | ALT | FA | |
| 1,5mg/dl | 0,4mg/dl | 1,1mg/dl | 22U/l | 18U/l | 77U/l | ||
| Estudios inmunológicos | C3 | C4 | ANA | ENA | Ac MPO | Ac PR3 | Coombs directo |
| 106mg/dl | 28,8mg/dl | Negativos | Negativos | Negativos | Negativos | Negativo | |
| Estudio genético del complemento | No se encontraron mutaciones patológicas en: ADAMTS13, C3, CD46, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, CFI, DGKE, PIGA, THBD | ||||||
| Otros estudios | Electroforesis de proteínas | VIH | Hepatitis B | Hepatitis C | VDRL | Biopsia renal | |
| Hipoalbuminemia, elevación inespecífica de la banda alfa 1 y B2 | Negativo | Negativo | Negativo | Negativo | Microangiopatía trombótica | ||
| TAC de cráneo simple | Ecocardiografía | Ecografía renal | |||||
| Normal | Hipertensión pulmonar y derrame pericárdico leve | Riñones pequeños, ecogénicos, con pérdida de la diferenciación corticomedular | |||||
Ac MPO: anticuerpos antimieloperoxidasa; Ac PR3: anticuerpos antiproteinasa 3; ALT: alanino aminotransferasa; ANA: anticuerpos antinucleares; AST: aspartato aminotransferasa; BD: bilirrubina directa; BI: bilirrubina indirecta; BT: bilirrubina total; BUN: nitrógeno ureico; CAP: campo de alto poder; Colesterol-LDL: colesterol de baja densidad; Colesterol-HDL: colesterol de alta densidad; C3: complemento C3; C4: complemento C4; ENA: anticuerpos contra los antígenos extraíbles del núcleo anti-RNP, Sm, Ro y La; ESP: extendido de sangre periférica; FA: fosfatasa alcalina; HCO3: bicarbonato sérico; LDH: lactato deshidrogenasa; PCO2: presión de dióxido de carbono; PO2: presión de oxígeno; PTH: paratohormona; TAC: tomografía axial computarizada; VIH: anticuerpo virus de la inmunodeficiencia humana; VRDL: prueba no treponémica para sífilis.
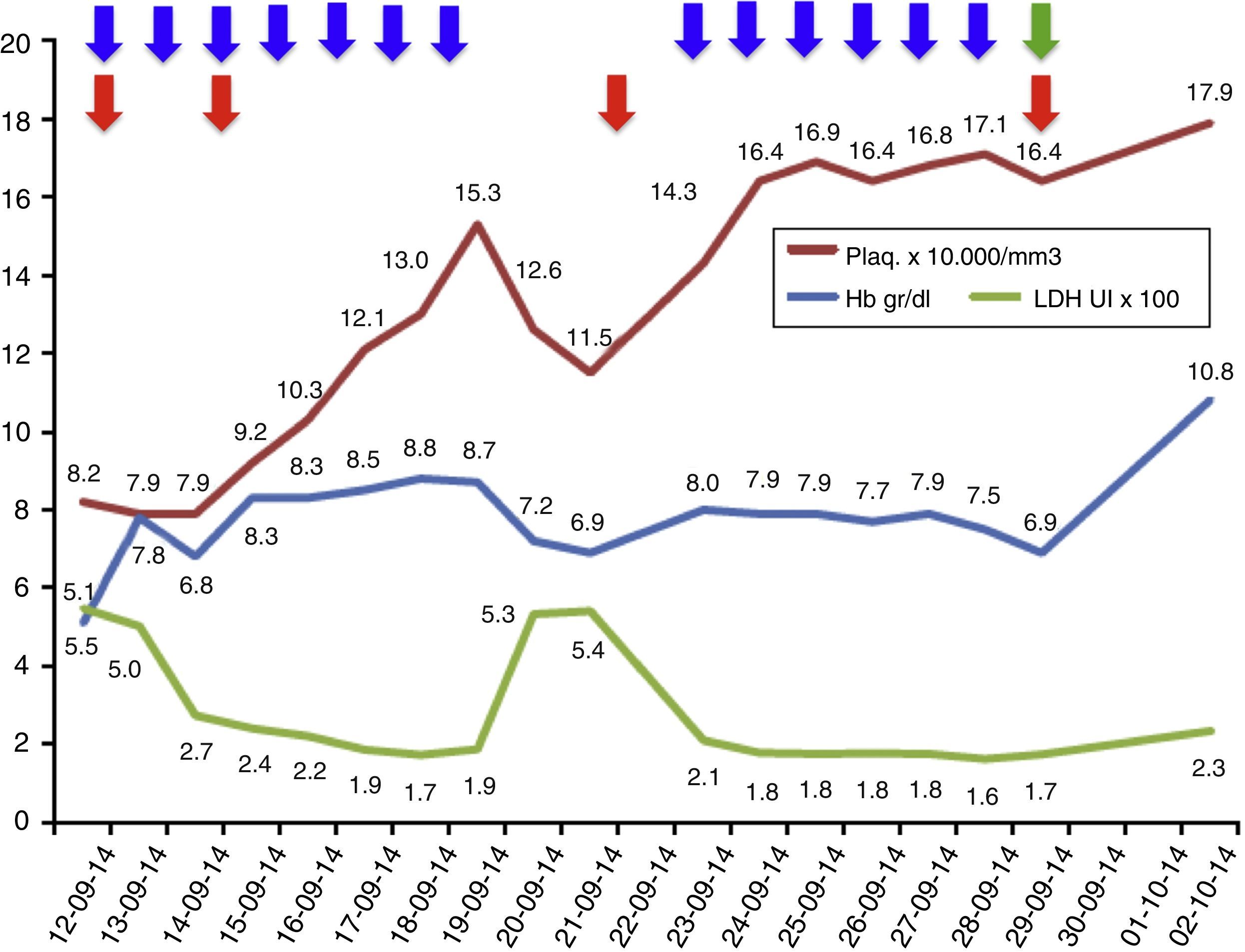
Evolución de los exámenes de laboratorio durante el tratamiento.
Evolución de los parámetros de laboratorio durante el tratamiento recibido: recambios plasmáticos 13 sesiones (flecha azules), transfusiones total 4 (flecha roja); además, se inició eculizumab el 29 de septiembre de 2014 (flecha verde). Los valores de LDH se encuentran en escala de 1×102 y los valores de plaquetas se encuentran en escala de 1×104.
El SHUa es una enfermedad genética crónica ultra-rara, ocasionada por una alteración en la regulación del complemento y que puede producir secuelas graves en múltiples órganos e incluso la muerte1–4. Se caracteriza por la tríada de anemia hemolítica microangiopática, trombocitopenia e insuficiencia renal, pero puede producir alteración en cualquier órgano1,2. Nuestro paciente presentó compromiso renal, hematológico, neurológico, cardiovascular y ocular, este último hallazgo, poco reportado en la literatura1,2.
En más de la mitad de los pacientes con SHUa se puede identificar la mutación del complemento o de otras moléculas que ocasiona la alteración en su regulación; sin embargo, no se requiere el estudio genético para confirmar el diagnóstico e iniciar el tratamiento, pero sí es ideal realizarlo para establecer el pronóstico, ya que hay algunas mutaciones más agresivas que otras; además, el estudio genético permite planear de una forma más adecuada el trasplante renal en los pacientes que lo requieren1,2,5. Hasta en el 30% de los enfermos no se documenta mutación4,6, como fue el caso de nuestro paciente.
El tratamiento de elección del SHUa es eculizumab, anticuerpo monoclonal humanizado que bloquea la escisión de la fracción C5 del complemento y, por lo tanto, evita la liberación de la anafilotoxina C5a y la formación del complejo de ataque de membrana C5b-9, evitando así el daño endotelial y la generación de microangiopatía trombótica1,7,8. Como prerrequisito para el inicio de eculizumab se debe aplicar la vacuna contra el meningococo e iniciar profilaxis antibiótica dirigida al meningococo, mientras la vacuna surte efecto. Cuanto más rápido se inicie eculizumab en los pacientes con SHUa, mayor será la posibilidad de recuperación y menores serán las secuelas5,9. Sin embargo, mientras se realiza el diagnóstico de este síndrome, los recambios plasmáticos pueden estabilizar la actividad hemolítica del paciente y, por lo tanto, se deben iniciar precozmente ante el cuadro clínico de una microangiopatía trombótica, teniendo en cuenta que se debe tomar previamente la actividad de la ADAMTS13 y el perfil de autoanticuerpos para no enmascarar otros diagnósticos1,5,7. Si al final el diagnóstico es un SHUa, el tratamiento debe ser cambiado a eculizumab, ya que la terapia plasmática a largo plazo no ha mostrado cambiar el curso devastador del SHUa1,5,9,10. En el caso de nuestro paciente, inicialmente se realizaron recambios plasmáticos, lográndose controlar la actividad hemolítica, y una vez realizamos el diagnóstico de SHUa, cambiamos el tratamiento a eculizumab, obteniendo buena respuesta, sin requerir nuevos recambios plasmáticos y sin presentar recaídas (fig. 1). Sin embargo, quedó como secuela ERCT.
El SHUa es una enfermedad genética ultra-rara que afecta a niños y adultos, caracterizada por la presencia de anemia hemolítica microangiopática no inmune, trombocitopenia y compromiso multiorgánico. Su curso clínico puede ser devastador y comprometer la vida o la integridad de los pacientes, dejando secuelas graves como la ERCT. El tratamiento con eculizumab debe ser iniciado precozmente para frenar el daño multiorgánico y evitar la muerte.
Conflicto de interesesEl Dr. Nieto-Ríos y la Dra. Serna-Higuita han dictado charlas sobre microangiopatía trombótica patrocinadas por Alexion Pharma. Los otros autores declaran no tener conflicto de interés con el contenido de este artículo.



