




El síndrome antisintetasa (SAS) es un trastorno no bien definido, poco frecuente, incluido hasta ahora dentro de las miopatías inflamatorias idiopáticas, que se caracterizan por presentar anticuerpos antisintetasa (ACAS) en el suero. Los ACAS son anticuerpos tipo IgG dirigidos contra la enzima sintetasa, que media la unión del ARN con un determinado aminoácido, para formar ARN transferente. De los principales ACAS descritos en la bibliografía, el primero y más frecuentemente (15-30%) encontrado es el anti-Jo1, que va dirigido contra la sintetasa que media la unión entre el ARN y la histidina1.
En la década de los 90 se empezaron a describir casos clínicos de pacientes con miositis, los ACAS en suero y algunas características clínicas más o menos comunes que vinieron a definirse SAS. Entre los criterios diagnósticos del SAS se incluyen: miopatía inflamatoria, que puede manifestarse clínicamente como debilidad muscular; enfermedad pulmonar intersticial que puede llegar a ocasionar un fallo respiratorio agudo con diferentes patrones histológicos (bronquiolitis obliterante, infiltrados intersticiales con predominio de células plasmáticas, y linfocitos y fibrosis pulmonar difusa); artritis inflamatoria no erosiva; las llamadas «manos de mecánico» que se tratan de lesiones hiperqueratósicas que se acompañan de fisuración, hiperpigmentación y escamas en la cara lateral y palmar de los dedos y palmas, y otros síntomas como fiebre, síndrome de Raynaud y síndrome seco1,2.
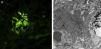
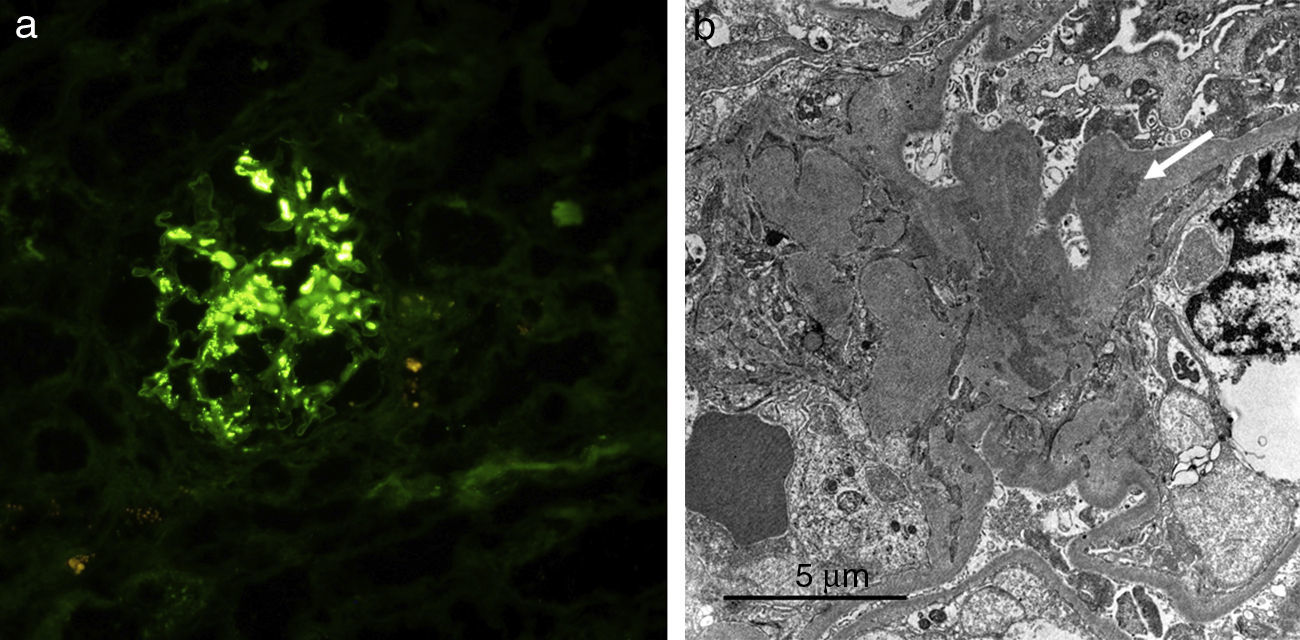
Presentamos el caso de un paciente de 59 años de edad, sin antecedentes de interés excepto ser fumador de unos 80 paquetes/año hasta 6 meses antes de su ingreso. En los 2 últimos meses desarrolló un cuadro de artralgias, mialgias y edemas en miembros superiores e inferiores acompañado de pérdida de fuerza. Coincidiendo con este cuadro presentó disnea de moderados esfuerzos. En la exploración física destacaba una presión arterial de 100/76mmHg, una saturación de oxígeno del 91% y ligeros edemas en ambos miembros inferiores. Entre las pruebas complementarias realizadas destacaba una gasometría arterial con un pH 7,47, pCO2 35mmHg, pO2 58mmHg, HCO3 25,5mmol/l, saturación de oxígeno 91,5%, hemoglobina 11,4g/dl, y leucocitos y plaquetas dentro de los rangos normales. En la bioquímica presentaba unas proteínas totales de 4,7g/dl, albúmina 2,74g/dl, GPT 414UI/l, GOT 436UI/l, GGT 11UI/l, LDH 1.443UI/l, CK 9.906UI/l, creatinina sérica 0,6mg/dl y urea 39mg/dl. Como reactantes de fase aguda resaltaba una velocidad de sedimentación globular de 37mmHg y una PCR 4,13mg/dl. En el estudio de autoinmunidad efectuado nos encontramos unos ANA-IFI negativos, un ANA-screening multiplex positivo, un Ac anti-Jo1/HRS positivo, y el resto del estudio incluido fue negativo con unos valores del complemento dentro de los rangos normales. En la orina mostraba la presencia de hematíes dismórficos en el sedimento urinario y una proteinuria de 0,4g/24h. Entre las pruebas de imagen sobresalía una radiografía de tórax con un patrón intersticial bilateral que se confirmaba en el escáner de tórax (hallazgos de afectación intersticial bilateral) y un electromiograma con signos de miopatía inflamatoria. Se realizó una biopsia muscular con hallazgos compatibles con miopatía inflamatoria, una miopatía inmune con patología perimisial (fig. 1a). En el estudio de inmunohistoquímica mostraba una sobreexpresión de MHC-I de forma difusa en la membrana de las fibras musculares (fig. 1b). Y una biopsia renal con un parénquima renal con leve expansión mesangial, y la presencia en la inmunofluorescencia directa depósitos de IgA (fig. 2a), y en la microscopía electrónica demostraba como principal alteración una discreta ampliación mesangial a partir de células como de matriz con frecuentes depósitos electrodensos homogéneos (fig. 2b). El paciente recibió tratamiento con esteroides a dosis de 1mg/kg/día y azatioprina 100mg/día con buena tolerancia y mejoría del cuadro clínico con descenso de los marcadores de daño muscular y disminución del patrón intersticial.
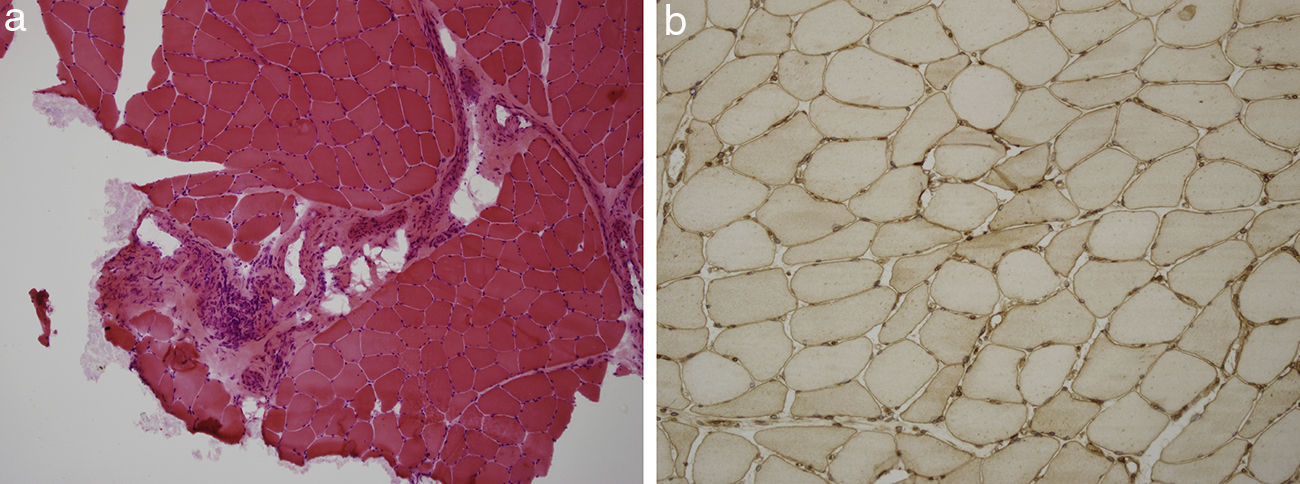
a). Estudio de hematoxilina-eosina de tejido músculo esquelético con infiltrado inflamatorio mononuclear de localización preferentemente perimisial. b). Estudio inmunohistoquímico de tejido muscular para MHC I (HLA) que muestra sobreexpresión del mismo en la membrana de las fibras musculares.
Aunque la presencia de los ACAS se ha considerado específica de la polimiositis y la dermatomiositis, desde hace unos años se ha ido perfilando un síndrome compuesto por la presencia de estos autoanticuerpos junto con enfermedad pulmonar intersticial, artritis, fiebre, lesiones hiperqueratósicas en las manos (manos de mecánico) y fenómeno de Raynaud, al que se ha llamado SAS. El cuadro clínico varía entre enfermedad pulmonar, muscular y/o articular, sin embargo los datos de afectación renal en esta entidad son realmente escasos3. Un excelente trabajo francés nos describe 14 casos de nefropatía y miopatía inflamatoria (documentada con biopsia), de los cuales 3 casos son un SAS4. Dos pacientes presentaban una nefropatía mesangial IgA y un caso se trataba de una nefropatía membranosa. Los 2 casos con nefropatía mesangial recibieron tratamiento inmunosupresor con esteroides y metotrexate consiguiendo una remisión completa de la enfermedad renal. La nefropatía mesangial IgA es la enfermedad glomerular asociada al SAS más documentada5–7. El pronóstico de estos pacientes suele venir determinado por la afectación pulmonar. El tratamiento óptimo del SAS se basa en la administración de esteroides e inmunosupresores, aunque existen casos descritos tratados con rituximab con éxito8.
Como conclusión, aunque las miopatías inflamatorias son unas conectivopatías poco frecuentes, debemos buscar la coexistencia con la enfermedad renal. La nefropatía glomerular más frecuentemente asociada en el SAS es la nefropatía mesangial IgA y presenta un curso favorable en la mayoría de los casos.



