



Anti-synthetase syndrome (ASS) is a poorly-defined, uncommon syndrome, and until recently was included within the idiopathic inflammatory myopathies, which are characterised by the presence of anti-synthetase antibodies (ASAb) in the serum. ASAbs are IgG antibodies directed against the enzyme synthetase, which mediates the binding of RNA to the specific amino acid, to form transfer RNA. Of the main ASAbs described in the bibliography, the first and most commonly found (15%–30%) is anti-Jo1, which is directed against the synthetase that mediates binding between RNA and histidine.1
During the 1990s, authors began to describe clinical cases of patients with myositis, the presence of ASAbs in serum, and some generally common clinical characteristics that came to be defined as ASS. AAS is characterized by the association of inflammatory myopathy, which is clinically as muscle weakness; interstitial lung disease, which can lead to acute respiratory failure of different histological patterns (bronchiolitis obliterans, interstitial infiltrates of predominantly plasma cells and lymphocytes, and diffuse pulmonary fibrosis); non-erosive inflammatory arthritis; the so-called mechanic's hands, which have hyperkeratotic lesions accompanied by fissures, hyperpigmentation, and scaling on the palm and on the palmar and lateral surfaces of the fingers; and other symptoms such as fever, Raynaud syndrome, and Sjögren syndrome.1,2
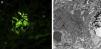
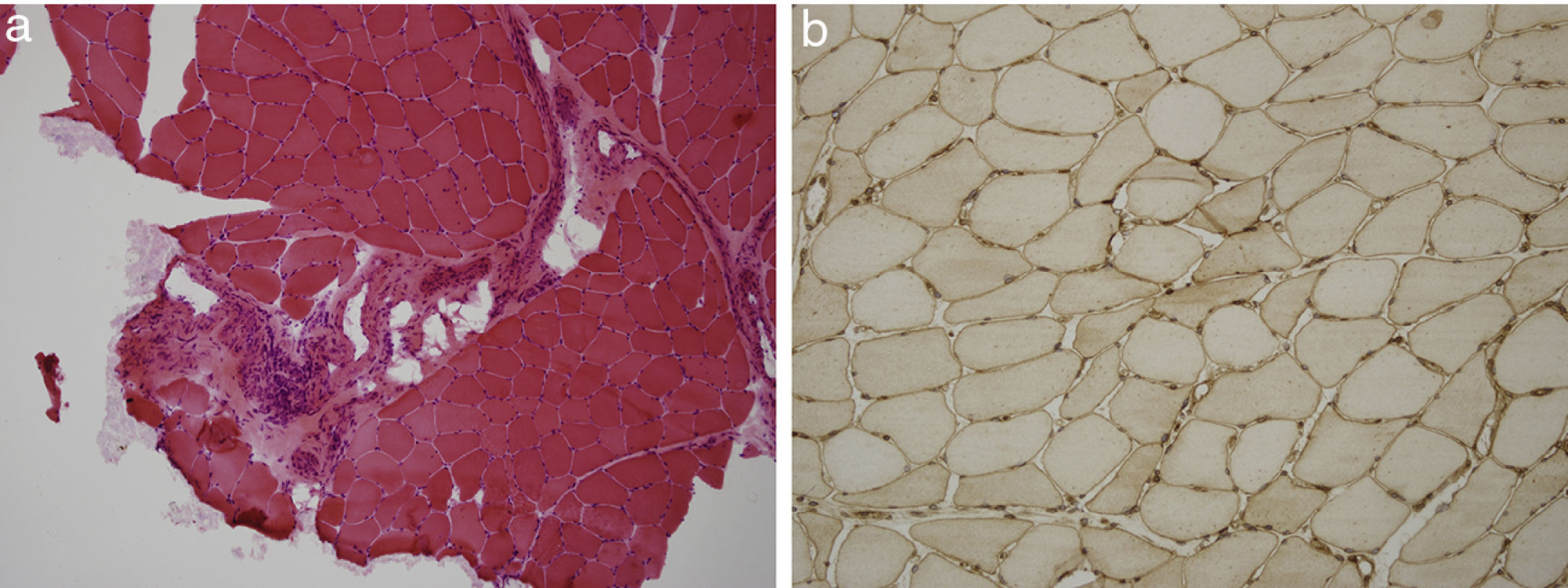
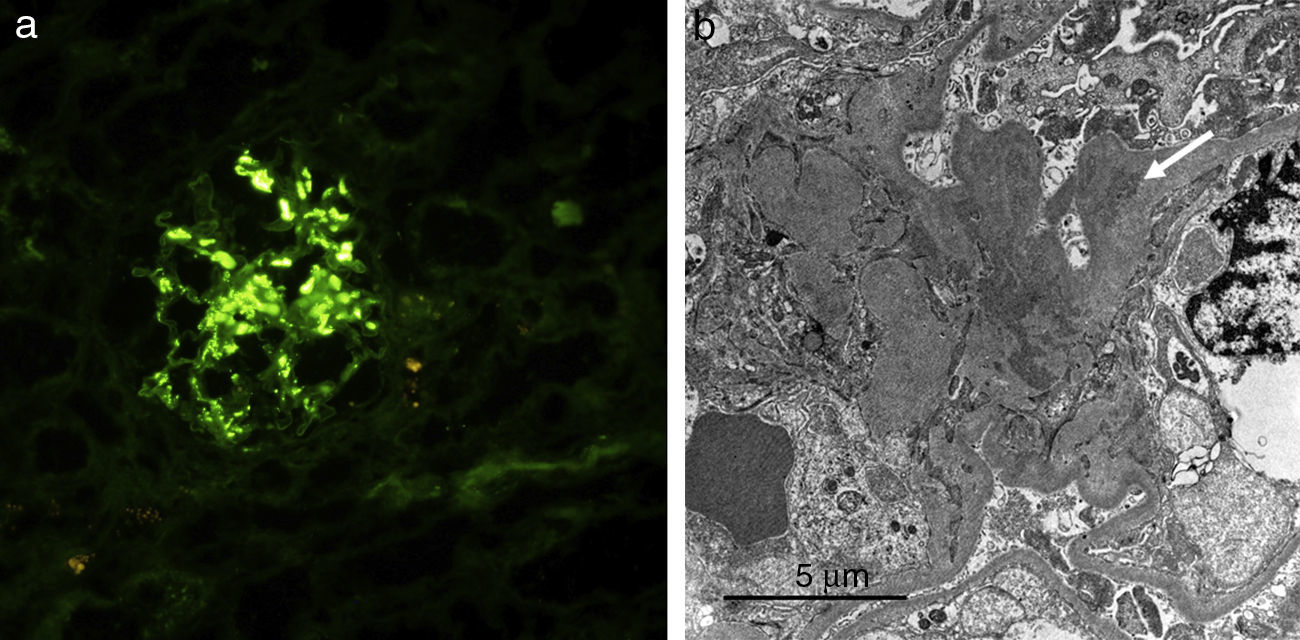
We present a case of a 59-year-old man, with no past medical history of note except for an 80 pack-year smoking history until 6 months before his admission. Over the previous 2 months he had developed symptoms of arthralgia, myalgia, and oedema in the upper and lower limbs, with associated loss of muscle strength. He also had moderate exertional dyspnoea. On physical examination he had a blood pressure of 100/76mmHg, oxygen saturation of 91% and mild oedema of both lower limbs. Investigations: arterial blood gas revealed a pH of 7.47, pCO2 35mmHg, pO2 58mmHg, HCO3 25.5mmol/L, oxygen saturation 91.5%. Haemoglobin was 11.4g/dL, and leukocytes and platelets were within normal limits. Biochemistry showed a total protein of 4.7g/dL, albumin 2.74g/dL, ALT 414UI/L, AST 436UI/L, GGT 11UI/L, LDH1.443UI/L, CK9.906UI/L, serum creatinine 0.6mg/dL, and urea 39mg/dL. Acute phase reactants included an erythrocyte sedimentation rate of 37mmHg and CRP of 4.13mg/dL. On the auto-immune study we found negative ANA-IFA, positive ANA-screening multiplex, a positive anti-Jo1/HRS Ab, and the rest of the study was negative, with complement values within normal limits. Urine contained dysmorphic erythrocytes in the urinary sediment and proteinuria of 0.4g/24h. Imaging included chest X-ray which showed a bilateral interstitial pattern confirmed on chest scan (findings of bilateral interstitial disease). Electromyography showed signs of inflammatory myopathy. The muscle biopsy findings were compatible with inflammatory myopathy, an immune myopathy with perimysial (around miocyte) pathology (Fig. 1a). On immunohistochemical study there was diffuse overexpression of MHC-1 in the membrane of the muscle fibres (Fig. 1b). Renal biopsy showed renal parenchyma with mild mesangial expansion. IgA was present on direct immunofluorescence (Fig. 2a). Electron microscopy showed, as the main abnormality, mild mesangial widening from both cells and matrix with frequent homogeneous electron-dense deposits (Fig. 2b). The patient received treatment with steroids at a dose of 1mg/kg/day and azathioprine 100mg/day, which was tolerated well, with clinical improvement, a reduction in markers of muscle damage, and resolution of the interstitial pattern.
Although the presence of ASAbs was previously considered specific to polymyositis and dermatomyositis, during the last few years there has been describing a syndrome comprising the presence of these autoantibodies along with interstitial lung disease, arthritis, fever, hyperkeratotic lesions on the hands (mechanic's hands) and Raynaud's phenomenon, which has been called ASS. The clinical presentation varies between pulmonary, muscle, and/or joint disease, but data on renal involvement in this condition are very scarce.3 One excellent French study described 14 cases of nephropathy and inflammatory myopathy (documented with biopsy), of which 3 cases were ASS.4 Two patients had mesangial IgA nephropathy and one patient had a membranous nephropathy. The 2 patients with mesangial nephropathy received immunosuppressive treatment with steroids and methotrexate, achieving complete remission of renal disease. Mesangial IgA nephropathy is the most documented glomerular disease associated with ASS.5–7 The prognosis of such patients is usually determined by pulmonary involvement. The optimal treatment for ASS is based on the administration of steroids and immunosuppressors, although there are other cases described that have been treated successfully with rituximab.8
In conclusion, although inflammatory myopathies are uncommon connective tissue diseases, we should look for their co-existence in patients with renal disease. In ASS, the most frequently-associated glomerular nephropathy is mesangial IgA nephropathy, which has a favourable outcome in most cases.
Please cite this article as: Morales E, Rabasco C, Panizo N, Gutierrez E, Martinez MA, Toldos O, et al. Nefropatía mesangial y síndrome antisintetasa: una forma curiosa de asociación. Nefrologia. 2015;35:415–417.



