



La enfermedad de Fabry es una enfermedad de depósito lisosomal de carácter hereditario, ligada al cromosoma X, causado por el déficit de la enzima alfa-galactosidasa A (alfa-GLA A), lo que conduce a la acumulación de glicoesfingolípidos, principalmente globotriaosilceramida, en los tejidos. Es una enfermedad poco prevalente, y con muy bajo índice de sospecha, por lo que, generalmente, existe un retraso en el diagnóstico y en el tratamiento.
Presentamos un caso de un paciente varón afecto de la enfermedad de Fabry, que presentaba múltiples quistes parapiélicos e insuficiencia renal, sin presentar angioqueratomas. El estudio genético mostró una mutación en el gen de alfa-GLA A que no ha sido descrita previamente en el registro de mutaciones, y de novo, ya que no se encontró en otros familiares, además que no fue trasmitida a la descendencia. La presencia de múltiples quistes parapiélicos y su peculiar aspecto fue lo que hizo sospechar el diagnóstico de la enfermedad.
Fabry disease is an inherited, X-linked lysosomal storage disorder caused by deficiency of the enzyme alpha galactosidase A (alpha-GLA A), which leads to glycosphingolipid accumulation, mainly globotriaosylceramide, in tissues. Disease prevalence and the index of suspicion are both low, which tends to result in delayed diagnosis and treatment.
We present the case of a male Fabry disease patient who manifested no angiokeratoma lesions but presented multiple parapelvic cysts and renal failure. The genetic study revealed an alpha-GLA A gene mutation that had not been recorded in the mutations registry. The de novo mutation was not found in his relatives and it was not transmitted to his offspring. The large number and peculiar appearance of the parapelvic cysts led to the diagnosis.
La enfermedad de Fabry (EF) es consecuencia de un déficit de la enzima alfa-galactosidasa A (alfa-GLA A), lo que conduce al depósito de globotriaosilceramida (Gb3) en los tejidos1. Es una enfermedad hereditaria, ligada al cromosoma X, que se manifiesta en los varones homocigotos y las mujeres heterocigóticas, y estas con afectación variable2. Es una enfermedad que puede aparecer en todas las razas, cuya prevalencia varía de 1:50.000 en varones3, sin embrago, en un estudio llevado a cabo con recién nacidos ha encontrado una incidencia de 1:37.0004. La EF tiene una amplia gama de manifestaciones clínicas, lo que hace que sea difícil establecer un diagnóstico oportuno. El inicio de los síntomas, por lo general, comienza en la infancia, y a menudo el dolor neuropático y múltiples angioqueratomas en la piel están presentes en un alto porcentaje de los casos5. Durante la edad adulta, empieza la afectación renal caracterizada por proteinuria con progresión a la insuficiencia renal crónica, la cual se asocia con un aumento en la mortalidad, en estos pacientes. Los quistes parapiélicos, también se han descrito en la EF como un hallazgo radiológico6.
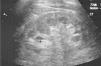
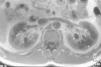
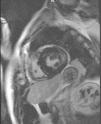
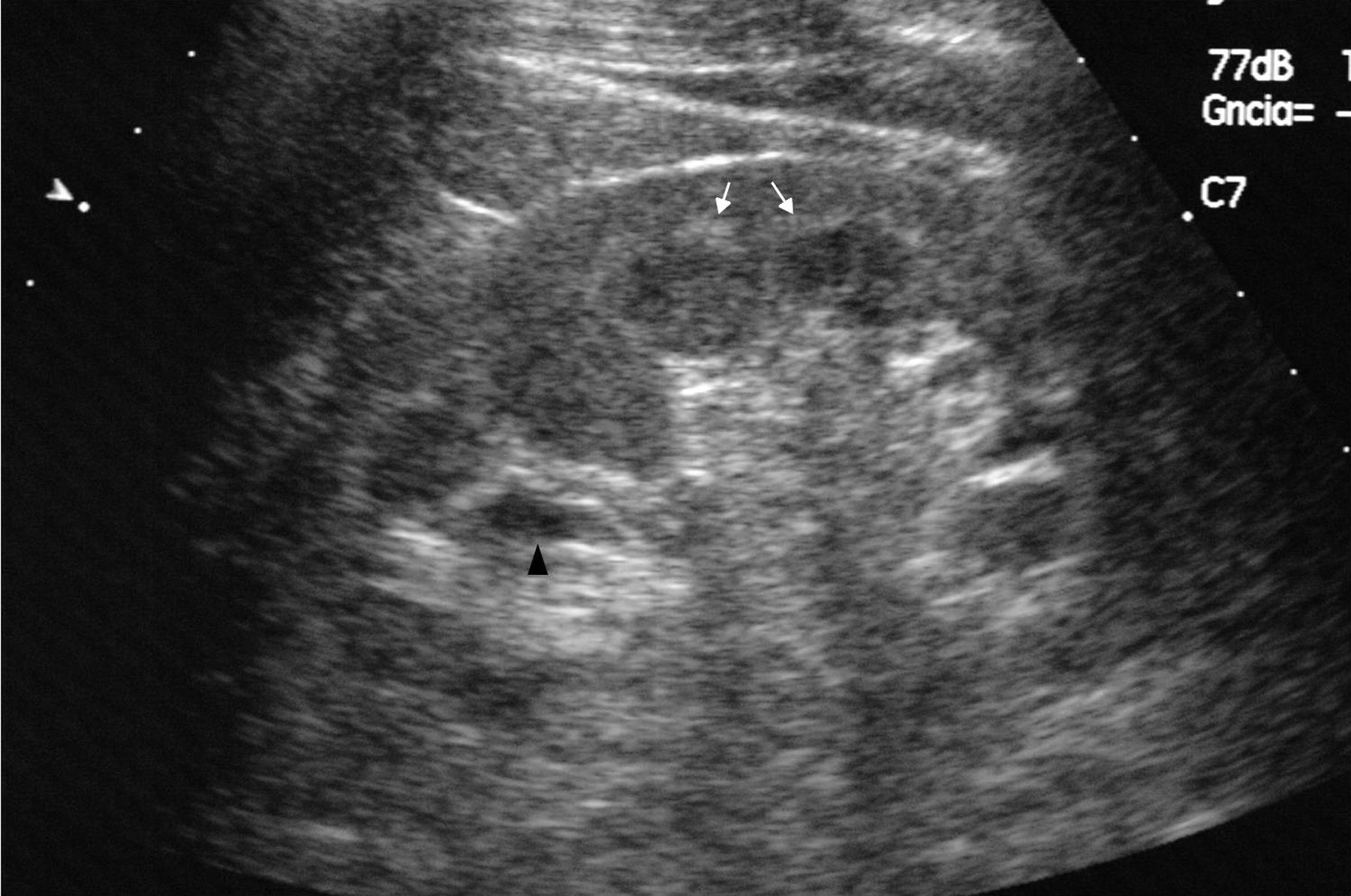
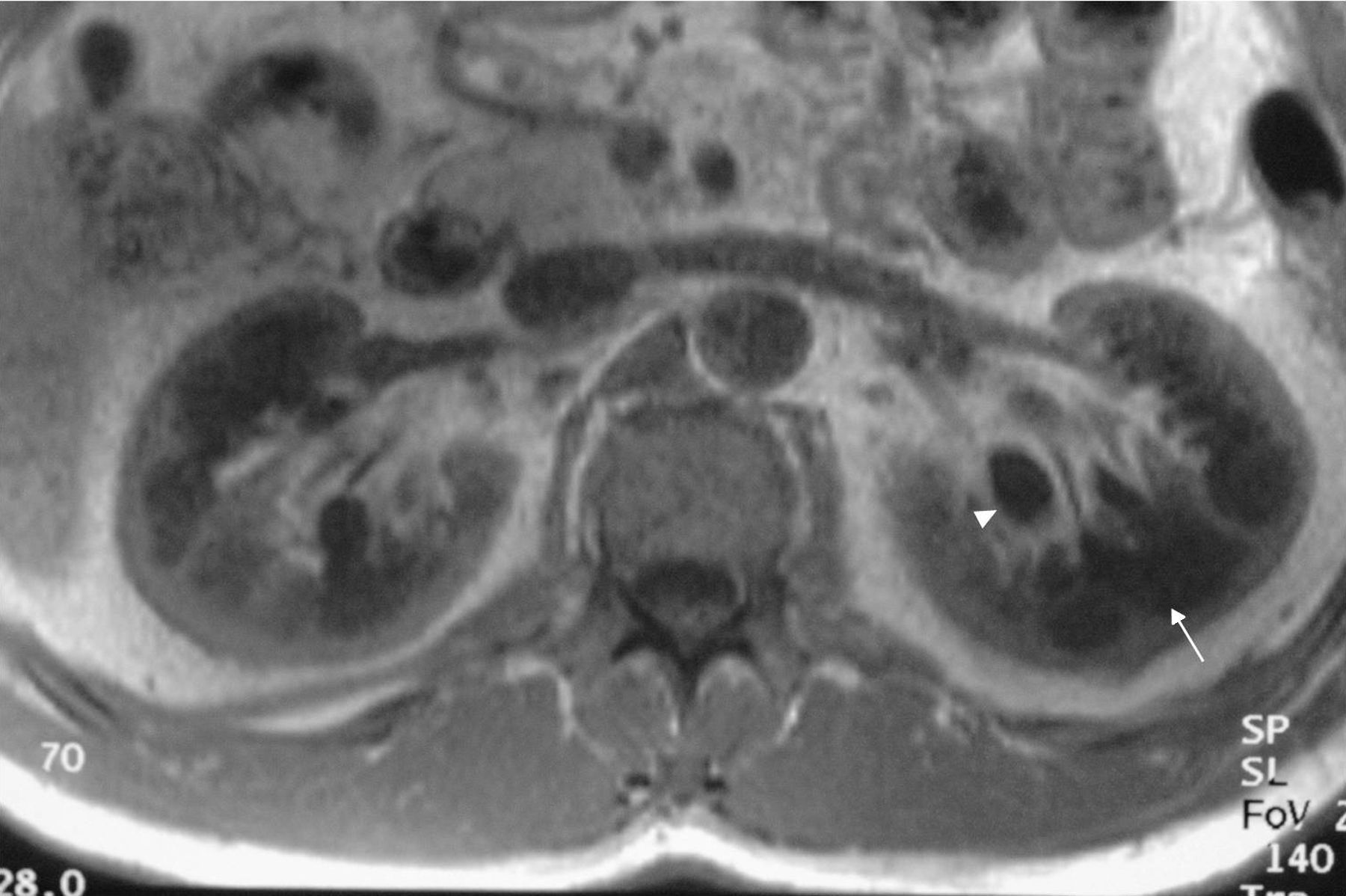
Caso clínicoPresentamos el caso de un varón de 47 años de edad, sin antecedentes familiares de EF que seguía controles en consulta externa de nuestro servicio desde el 2002 por proteinuria. En 2006, la ecografía abdominal y la resonancia magnética (RM) mostraron una mala diferenciación córtico-medular y la presencia de múltiples quistes parapiélicos bilaterales (figs. 1 y 2). En 2007, por deterioro de la función renal con niveles de creatinina sérica de 1,6mg/dl y proteinuria de 1g/24h, se decidió realizar una biopsia renal guiada por ecografía, que reveló cambios consistentes con moderada-grave nefritis intersticial crónica. En 2009, el paciente ingresó debido a edemas y disnea de esfuerzo. La exploración no mostró lesiones cutáneas sugestivas de angioqueratomas en piel. La analítica de ingreso mostró una creatinina sérica de 1,7mg/dl y proteinuria de 2,4g/24h. Los hallazgos en el ecocardiograma y en la RM cardiaca mostraron una probable miocardiopatía por enfermedad de depósito (fig. 3). La electromiografía mostró signos de neuropatía de fibras pequeñas, con alteraciones de la función autonómica. Se realizó una prueba de sudor que no recogió sudor tras la estimulación con pilocarpina, y un estudio oftalmológico que reveló córnea verticillata.

Ante la sospecha clínica de EF, se determinó el nivel de actividad de la alfa galactosidasa en leucocitos y plasma, utilizando un sustrato fluorogénico 4-methyilumbelliferyl α-D-galactosidasa. La actividad GLA era 0,5nmol/h/ml (2% de lo normal) en el plasma, y /4,2nmol/h/mg de proteína (10,7% de lo normal) en los leucocitos, lo que confirma el diagnóstico de la EF. La actividad de la enzima fue normal en los familiares estudiados del paciente (madre y hermana). Se realizó el estudio genético, el ácido desoxirribonucleico genómico (ADN) se extrajo de mancha de sangre seca y los fragmentos de genes de codificación se amplificaron mediante PCR. La secuenciación directa de los 7 exones del gen de GLA se realizó usando un secuenciador capilar (ABI PRISM® 310 Genetic Analyzer). El análisis determinó que el paciente era homocigoto para una nueva mutación c.1182del (delección de un solo nucleótido) en el exón 7 del gen alfa-GLA A. Se investigó a la madre y hermana del paciente, siendo negativas para esta mutación. Los hermanos restantes no estaban disponibles para el estudio, pero ninguno de ellos ha manifestado síntomas clínicos asociados con la enfermedad, hasta la fecha. El paciente tiene un hijo varón, por lo que la mutación no se transmite a la descendencia. Se inició la terapia de reemplazo hormonal con agalsidasa alfa (Replagal®). Durante el seguimiento, el edema y el dolor neuropático mejoraron, pero la función renal empeoró progresivamente hasta la insuficiencia renal terminal, en un período de 3 años. El paciente inició hemodiálisis, y un año más tarde, recibió un injerto renal de donante vivo de su esposa. El paciente, durante el seguimiento postrasplante, ha permanecido con función renal estable con un filtrado glomerular estimado de 45ml/min/1,73m2, con proteinuria de 200mg/24h. Se han realizado biopsias de seguimiento a los 3 meses y al año, que no han mostrado depósitos de Gb3 en el tejido renal. En cuanto a la evolución cardiaca, clínicamente ha presentado aparición de disnea de esfuerzo, además de empeoramiento de la fibrosis miocárdica, valorada por resonancia cardiaca realizada en febrero de 2014. Se decidió cambio de tratamiento a agalsidasa beta (Fabrazyme®), desde entonces.
DiscusiónLos quistes parapiélicos no son infrecuentes en la EF, pero no son específicos para esta condición. En un estudio anterior de 122 EF pacientes (76 varones y 40 mujeres), se observó un aumento en la frecuencia, de quistes corticales y parapiélicos, en relación a la población general6. Otro estudio encontró que los quistes renales parapiélicos fueron más frecuentes en los pacientes con EF que en los controles sanos7. Aunque nuestro paciente no mostró angioqueratomas, una de las manifestaciones clínicas más frecuentes en los pacientes con EF, el cuadro clínico y los hallazgos en los estudios de imagen nos llevaron a sospechar una base genética para la enfermedad. El hallazgo de múltiples quistes del seno renal en paciente joven con insuficiencia renal de causa desconocida, debe plantear la posibilidad de la EF en el contexto clínico apropiado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



