


Hypokalaemia is a common clinical problem. A potential but commonly overlooked cause of hypokalaemia is Gitelman syndrome.
Material and methodsA 26-year-old man was admitted to the hospital due to syncope with general and muscular weakness and muscle cramps. The patient's history revealed previous recurrent syncope events associated to hypokalaemia with the lowest serum potassium value being 2.6mmol/l. At admission, blood pressure was normal and no changes were found at physical examination. Laboratory tests showed mild hypokalaemia (3.0mmol/l), hypomagnesaemia (1.36mg/dl), hypocalciuria (< 40mg/24h), and metabolic alkalosis (HCO3− 29.7mmol/l, BE 5.3mmol/l).
ResultsFurther laboratory tests (FeK, TTKG) confirmed inappropriate kaliuresis. Conn's disease was excluded by hormonal and imaging assessments. Genetic testing was performed and two novel heterozygous mutations: c.35_36insA and c.1095+5G>A were found in transcript NM_000339.2 in SLC12A3 gene.
ConclusionThe patient was diagnosed with Gitelman syndrome and was treated with supplements of potassium and magnesium.
La hipopotasemia es un problema clínico común. El síndrome de Gitelman es una posible causa de hipopotasemia a veces no reconocida.
Material y métodosUn hombre de 26 años de edad ingresa en un hospital por causa de un síncope, debilidad generalizada y calambres musculares. La historia clínica del paciente reveló la incidencia del síncope con hipopotasemia recurrente con el valor más bajo de potasio en 2,6mmol/l. En el ingreso, el paciente presentaba una presión arterial normal y la exploración física no reveló ninguna enfermedad. La evaluación del laboratorio demostró una hipopotasemia leve (K+ 3,0mmol/l), hipomagnesemia (Mg 1,36mg/dl), hipocalciuria (<40mg/24h) y alcalosis metabólica (HCO3- 29,7mmol/l, exceso de base 5,3mmol/l).
ResultadosOtras pruebas de laboratorio (FeK, TTKG) confirman una caliuresis inadecuada. La enfermedad de Conn fue excluida tras la evaluación hormonal y radiológica. Se realizaron las pruebas genéticas y 2 mutaciones heterocigóticas: c.35_36insA y c.1095+5G>A fueron encontradas en la transcripción NM_000339.2 del gen SLC12A3.
ConclusiónEl paciente fue diagnosticado con el síndrome de Gitelman y fue tratado con suplementos de potasio y magnesio.
Hypokalemia is a common clinical problem in endocrinologists’ and nephrologists’ practice. There are many obvious causes of hypokalemia such as diarrhea, vomiting or diuretics abuse. Other causes such as tubulopathies are rarely observed and their diagnosis is more challenging. There are many inherited and acquired tubulopathies causing hypokalemia, sometimes severe and life-threatening.1
A relatively common but overlooked cause of hypokalemia is Gitelman syndrome (GS).2 It is a recessive salt-losing tubulopathy caused by the SLC12A3 gene mutation. SLC12A3 gene encodes the thiazide-sensitive transporter NCCT (sodium chloride co-transporter). NCCT is located in the distal convoluted tubular cells (DCC), which are responsible for 7–10% of electrolyte tubular absorption.3
The most severe laboratory abnormalities found in GS are hypokalemia and hypomagnesaemia caused by renal K+ and Mg2+ wasting. Other typical changes are metabolic alkalosis, hypocalciuria and hyperreninemic hyperaldosteronism.4 Mild to moderate hypophosphatemia is frequently observed.5 Severe hypophosphatemia with severe hyponatremia was also reported.6,7
First symptoms of GS occur in children or young adults with normal growth and history of salt-craving behaviors (children eager to consume pickle or brine, salted cucumbers, oranges and lemons, children licking salt from potato crisps, etc.).8 Clinical presentation varies among patients. Some are asymptomatic but others develop life-threatening complications. Males manifest a more severe phenotype than females.8 The most common symptoms are muscular cramps and weakness, constipation, nocturia, polyuria, thirst, polydipsia, cardiac arrhythmias, paresthesias and increased salt appetite. Arterial hypotension is common and in many cases the most prominent symptom, however, in aging GS population hypertension can occur.8 The correlation between biochemical abnormalities and symptoms is not strong.9 GS does not interfere with children's moods and social relationships.9,10 Otherwise symptoms are more common in adults and can have negative impact on their quality of life. Forty-five percent of GS patients consider their symptoms as a moderate to big problem.11 Extreme exhaustion, muscular weakness, paresthesias, severe fatigue and hypotension are associated with mild to severe reduction in daily activities.9
Estimated prevalence of GS is 1:40,0008 and the prevalence of heterozygous is at least 1% in the European population. More than 180 different mutations in SLC12A3 have been described until now.12
Case reportA 26-year-old male was admitted to the hospital due to incidence of syncope, generalized and muscular weakness and muscle cramps. The patient's history revealed an episode of syncope with potassium level 3.16mmol/l. In further follow up in outpatient assessment, recurrent incidence of hypokalemia (the lowest value 2.6mmol/l) was observed. Blood pressure was normal 110/80, heart rate was 72 per minute; there was no changes in physical examination. Nor neurological findings, weight 74kg, height 178cm. On admission to hospital laboratory evaluation showed mild hypokalemia (K+ 3.0mmol/l), hypomagnesaemia (Mg2+ 1.36mg/dl), hypocalciuria (<40mg/24h), and metabolic alkalosis (HCO3− 29.7mmol/l, BE 5.3mmol/l). Kidney function was good with eGFR>60ml/min. Imaging studies were unremarkable, so was the ECG.
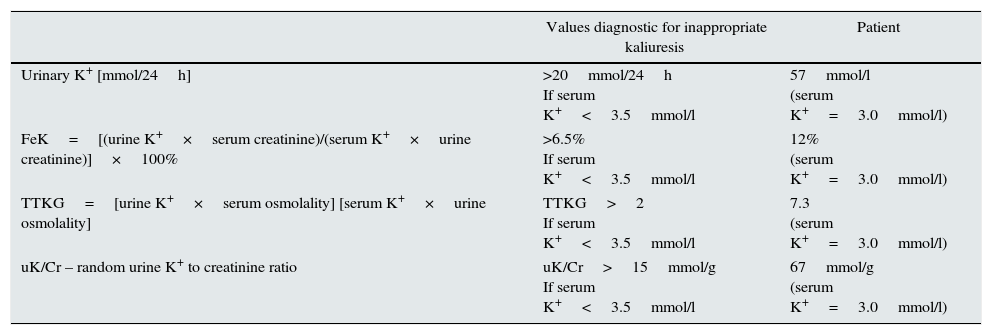
Further investigations confirmed that hypokalemia was caused by renal potassium wasting. The 24-h potassium wasting, transtubular K+ gradient (TTKG), fractional K+ excretion (FeK) and random K/creatinine ratio (K/Cr) were typical for hyperkaliuria and are shown in Table 1.13–15 Daily magnesiuria was 64mg/24h, with fractional magnesium (FeMg) excretion 15%.
The values diagnostic for hypokalemia.13–15
| Values diagnostic for inappropriate kaliuresis | Patient | |
|---|---|---|
| Urinary K+ [mmol/24h] | >20mmol/24h If serum K+<3.5mmol/l | 57mmol/l (serum K+=3.0mmol/l) |
| FeK=[(urine K+×serum creatinine)/(serum K+×urine creatinine)]×100% | >6.5% If serum K+<3.5mmol/l | 12% (serum K+=3.0mmol/l) |
| TTKG=[urine K+×serum osmolality] [serum K+×urine osmolality] | TTKG>2 If serum K+<3.5mmol/l | 7.3 (serum K+=3.0mmol/l) |
| uK/Cr – random urine K+ to creatinine ratio | uK/Cr>15mmol/g If serum K+<3.5mmol/l | 67mmol/g (serum K+=3.0mmol/l) |
Although blood pressure was normal, hormonal tests to exclude Conn's disease and the abdominal CT were performed, and did not reveal any abnormalities in adrenal glands. Secondary hyperaldosteronism with levels of aldosterone 289pg/ml (normal range 20–180) and renin 205mIU/ml (normal range 2.8–39.9) were typical for GS.
The patient was diagnosed as GS on the basis of a clinical phenotype. After the diagnosis, treatment with supplements of potassium and magnesium was introduced. He received 20mmol of potassium chloride and 18mmol of magnesium pyrrolidone carboxylate, with clinical improvement. Family history revealed mild asymptomatic hypokalemia in the patient's 35-year-old sister. But investigations showed neither hyperkaliuria nor hypomagnesaemia. GS was not diagnosed in this case.
Genetic analysisAccording to the genetic analysis, algorithm proposed by Nozu16SLC12A3 gene should be tested in patients with hypokalemic metabolic alkalosis, with full term birth, normal weight without nephrocalcinosis, with hypocalcuria and hypomagnesaemia.
In the described case genetic testing was performed and two heterozygous mutations: c.35_36insA and c.1095+5G>A were found in transcript NM_000339.2 of the SLC12A3 gene (Fig. 1). Both mutations are not yet in the HGMD(r) database version available [HGMD® Professional 2013.2 – 28th June 2013]. The first mutation was also found in patient's mother and the second in father. Only one of the two mutations identified in our patient c.35_36insA was found in his sister.

The mutations found in this patient almost prove the clinical diagnosis of GS. The pathogenetic relevance of the frameshift mutation is obvious. We cannot be sure about the pathogenetic relevance of the splice site mutation of intron 8. The consensus splice sequence G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) at the splice donor of intron 8 does not exactly match the consensus GTgAGc. The mutation found in this patient further deviates the sequence from the consensus (GtgAac). Also, in silico analysis by mutation taster considers this mutation as disease causing.17
DiscussionTubulopathies are rare diseases. According to RenalTube database18 the most common primary tubulopathies in European population are distal renal tubular acidosis, Bartter syndrome, familial hypomagnesaemia with hypercalciuria and GS.18 The prevalence of GS is around 25 cases per 1 million.3 Because GS is one of the most common, probably many nephrologists and endocrinologist will be confronted with cases of GS during their careers. Our patient had typical clinical presentation of GS and responded well to therapy. The problem with GS is that overlooked hypokalemia could cause death due to cardiac arrest or respiratory muscles paralysis. The severe neuromuscular symptom such as hypokalemic paralysis occurs in up to 6% of patients (more common in Asian patients).3
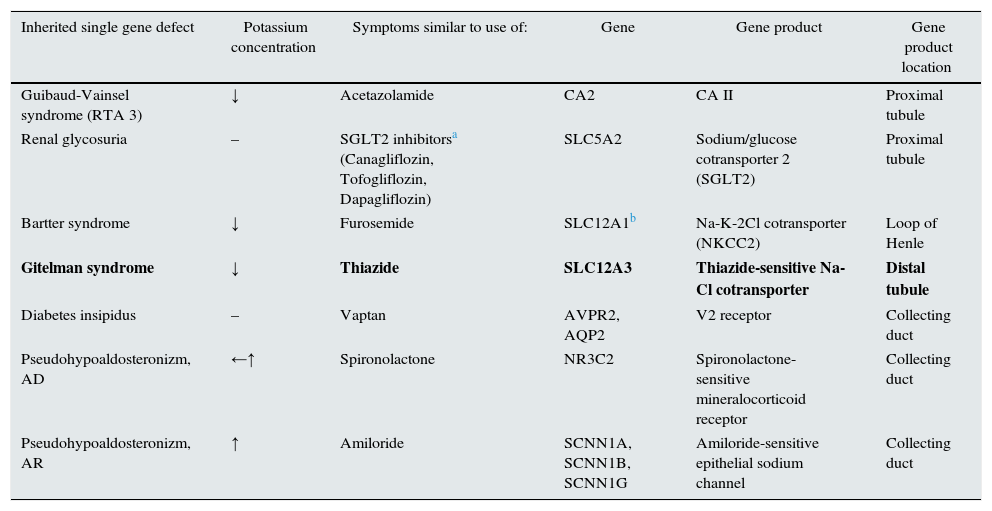
The inherited tubulopathies have a kind of “mirror images”-acquired tubulopathies caused by diuretics. Every diuretic (to be precise: natriuretics, acquaretics or glucuretics) cause abnormalities similar to those found in inherited tubulopathies (Table 2). Almost in every tubulopathy polyuria occurs, and potassium level changes are among the commonest problems. In patients with GS the mutations of SLC12A3 gene are found. This gene is encoding the thiazide-sensitive transporter. Therefore GS resembles chronic treatment with thiazides.
Diuretics and tubulopathies.
| Inherited single gene defect | Potassium concentration | Symptoms similar to use of: | Gene | Gene product | Gene product location |
|---|---|---|---|---|---|
| Guibaud-Vainsel syndrome (RTA 3) | ↓ | Acetazolamide | CA2 | CA II | Proximal tubule |
| Renal glycosuria | – | SGLT2 inhibitorsa (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Sodium/glucose cotransporter 2 (SGLT2) | Proximal tubule |
| Bartter syndrome | ↓ | Furosemide | SLC12A1b | Na-K-2Cl cotransporter (NKCC2) | Loop of Henle |
| Gitelman syndrome | ↓ | Thiazide | SLC12A3 | Thiazide-sensitive Na-Cl cotransporter | Distal tubule |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | V2 receptor | Collecting duct |
| Pseudohypoaldosteronizm, AD | ←↑ | Spironolactone | NR3C2 | Spironolactone-sensitive mineralocorticoid receptor | Collecting duct |
| Pseudohypoaldosteronizm, AR | ↑ | Amiloride | SCNN1A, SCNN1B, SCNN1G | Amiloride-sensitive epithelial sodium channel | Collecting duct |
AR – autosomal recessive; AD – autosomal dominant.
GS is also known as familial hypokalemic hypomagnesaemia, because hypokalemia is the most common phenomenon. In a patient with hypokalemia and suspicion of tubulopathy one should confirm hyperkaliuria. Renal potassium wasting can be proved by calculating TKKG, FeK, random K/Cr ratio or after 24-h urine collection (Table 1). Assessment of kaliuria should be performed, when the patient is taking neither diuretics nor potassium supplementation and when the potassium level is low. When potassium excretion is below 30mmol/l and TTKG is low, hypokalemia is caused by extrarenal loss or a transcellular K+ shift.
When excessive potassium renal loss in patient with hypokalemia is confirmed, further diagnosis is based on pH and blood pressure. In many cases these simple studies will be enough for diagnosis. An important problem of an increasing interest is primary aldosteronism (PA). Some clinical studies have suggested that PA is the cause of over 10% of arterial hypertension (AH), and is more common in patients with AH resistant to antihypertensive agents.19 Hypokalemia is one of the “classical” symptoms of PA, but is not so common as was considered.20 Yet, because there is no gold standard in the diagnosis of AP, it is worth remembering about RAA axis in patients with AH and hypokalemia.19
Diagnosis of hypomagnesaemiaHypomagnesaemia is a common abnormality in GS patients, although it is not observed in every case.21 In normomagnesemic patients with GS, clinical manifestation and electrolyte abnormalities are milder.22
Similar to potassium ions, magnesium ones are freely filtered by the glomeruli. 10% of filtered Mg2+ is absorbed by the proximal tubule, 50–70% in ascending limb of Henle's loop. Distal reabsorption depends on epithelial Mg2+ TRPM6 channels. Magnesiuria depends on oral intake and FeMg should be calculated to establish renal Mg2+ wasting (FeMg=(urine magnesium×serum creatinine)/[0.7(serum magnesium×urine creatinine)]×100). FeMg less than 2% suggests poor intake, GI losses or shift of Mg into cells. FeMg above 4% suggests renal Mg2+ wasting. When renal Mg wasting is proved, random Ca/Cr ratio should be calculated. Urine Ca/Cr ratio <0.3 is typical in GS, isolated dominant hypomagnesaemia with hypocalciuria and in thiazide treatment. Urine Ca/Cr ratio >0.3 is typical in hypermagesuria in Bartter syndrome, so are isolated recessive hypomagnesaemia with normocalciuria, familial hypomagnesaemia with hypercalciuria and nephrocalcinosis, autosomal dominant hypocalcemia with hypercalciuria, loop diuretics treatment and nephropathy caused by some nephrotoxins.23
Magnesium and potassium homeostasis are related to each other, and potassium depletion cannot be corrected until the correction of hypomagnesaemia.23 On average patients should receive up to 500mEq of potassium, 4–5mg/kg/day of 5–10mg of magnesium chloride. Amiloride (5–10mg/day) and spironolacton (200–300mg) are helpful.3
Clinical symptoms and laboratory results are essential for diagnosis. In cases with typical phenotype some authors recommend performing a thiazide test to confirm diagnosis.24 In presented case genetic investigation revealed two novel mutations of SLC12A3 gene. They are probably caused by lack of functional mutations of NCCT.
ConclusionsGS syndrome is one of the rare causes of hypokalemia, and it seems a challenge for physicians. We show in this paper that if one remembers about a very simple approach to hypokalemia and is aware of diuretics action and their similarity to inherited tubulopathies, the diagnosis could be quite straightforward. GS should be differentiated from other tubulopathies (inherited as well as acquired), and other causes of hypokalemia (e.g. Conn's disease). Familial history can reveal asymptomatic patients with GS. Suitable treatment protects patients from potentially dangerous complications.



