












La cistinosis es una enfermedad lisosomal minoritaria de expresión sistémica con especial afectación renal y oftalmológica, en la que los pacientes inician terapia renal sustitutiva en la primera década de la vida en ausencia de tratamiento. El pronóstico de la cistinosis depende del diagnóstico precoz, la pronta instauración del tratamiento con cisteamina y el buen cumplimiento terapéutico. La progresión de la enfermedad renal y de las complicaciones extrarrenales y una menor supervivencia, son más acentuadas en pacientes no adherentes.
ObjetivoEl objetivo de este trabajo fue la elaboración de unas recomendaciones para la atención integral de la cistinosis y la transición del adolescente a la medicina del adulto, basadas en la experiencia clínica, con el fin de reducir el impacto de la enfermedad y mejorar la calidad de vida y el pronóstico del paciente.
MétodoBúsqueda bibliográfica y reuniones de consenso de un equipo multidisciplinar de expertos en la práctica clínica con pacientes afectos de cistinosis (Grupo T-CiS.bcn), procedentes de 5 hospitales localizados en Barcelona.
ResultadosEl documento recoge recomendaciones específicas y necesarias para el diagnóstico, tratamiento y seguimiento multidisciplinar de la cistinosis en las siguientes áreas: nefrología, diálisis, trasplante renal, oftalmología, endocrinología, neurología, laboratorio, consejo genético, enfermería y farmacia.
ConclusionesDisponer de un documento de referencia para la atención integral de la cistinosis constituye una herramienta de soporte para los profesionales de la salud que asisten a estos pacientes. Los principales pilares en los que se sustenta son: a) el enfoque multidisciplinar, b) la adecuada monitorización de la enfermedad y control de los niveles de cistina intraleucocitarios, c) la importancia de la adherencia al tratamiento con cisteamina y d) la promoción del autocuidado del paciente mediante programas de educación en la enfermedad. Todo ello conducirá, en una segunda fase, a la elaboración de un modelo de transición coordinado entre los servicios de pediatría y de adultos que contemple las necesidades específicas de la cistinosis.
Cystinosis is a rare lysosomal systemic disease that mainly affects the kidney and the eye. Patients with cystinosis begin renal replacement therapy during the first decade of life in absence of treatment. Prognosis of cystinosis depends on early diagnosis, and prompt starting and good compliance with cysteamine treatment. Kidney disease progression, extra-renal complications and shorter life expectancy are more pronounced in those patients that do not follow treatment.
The objective of this work was to elaborate recommendations for the comprehensive care of cystinosis and the facilitation of patient transition from paediatric to adult treatment, based on clinical experience. The goal is to reduce the impact of the disease, and to improve patient quality of life and prognosis.
MethodsBibliographic research and consensus meetings among a multidisciplinary professional team of experts in the clinical practice, with cystinotic patients (T-CiS.bcn group) from 5 hospitals located in Barcelona.
ResultsThis document gathers specific recommendations for diagnosis, treatment and multidisciplinary follow-up of cystinotic patients in the following areas: nephrology, dialysis, renal transplant, ophthalmology, endocrinology, neurology, laboratory, genetic counselling, nursing and pharmacy.
ConclusionsA reference document for the comprehensive care of cystinosis represents a support tool for health professionals who take care of these patients. It is based on the following main pillars: a) a multi-disciplinary approach, b) appropriate disease monitoring and control of intracellular cystine levels in leukocytes, c) the importance of adherence to treatment with cysteamine, and d) the promotion of patient self-care by means of disease education programmes. All these recommendations will lead us, in a second phase, to create a coordinated transition model between paediatric and adult care services which will cover the specific needs of cystinosis.
La cistinosis es una enfermedad lisosomal minoritaria de expresión sistémica que, en ausencia de tratamiento, conduce al fallo renal terminal en la primera década de la vida1. Su historia natural se ha transformado gracias al desarrollo del trasplante renal (TxR) en niños2 y la disponibilidad del tratamiento específico con cisteamina, fármaco que debe mantenerse de por vida3. Como consecuencia, la supervivencia de los pacientes ha aumentado desde la primera hasta más allá de la cuarta década de la vida, de modo que la cistinosis ha trascendido del ámbito pediátrico a la medicina del adulto4.
El control de la cistinosis es complejo por su gravedad, su naturaleza multisistémica y por precisar un tratamiento con múltiples fármacos de régimen posológico muy estricto. El diagnóstico precoz, la administración temprana de cisteamina y la adherencia al tratamiento, condicionan la morbilidad y el pronóstico vital5,6. Pese a ello, el cumplimiento terapéutico, que suele ser adecuado en el niño, tiende a disminuir en el adolescente y en el adulto7. Paralelamente, cuando el paciente alcanza la edad adulta, suele ser transferido desde el centro experto pediátrico hacia el hospital local con experiencia limitada en cistinosis, a la par que progresan las manifestaciones sistémicas y la complejidad de la enfermedad8. Este fenómeno es conocido en otras patologías renales crónicas de debut pediátrico9 y subraya la necesidad de implementar estrategias de transición y promover el autocuidado del paciente10.
El mapa actual de la cistinosis en España lo componen 56 pacientes atendidos en 22 centros hospitalarios. De ellos, aproximadamente un 50% son adultos y un 16% adolescentes. El 57% son trasplantados renales7. El proyecto del grupo de trabajo para la transición en cistinosis de Barcelona (T-CiS.bcn) reúne un grupo de expertos en la enfermedad con el objetivo de elaborar unas recomendaciones dirigidas a la atención integral de la cistinosis y la transición del adolescente a los servicios de adultos. El documento que presentamos constituye una herramienta de soporte para los profesionales de la salud que asisten a estos pacientes con cistinosis, enfocado a reducir el impacto de la enfermedad, mejorar la calidad de vida y aumentar la supervivencia, siguiendo las directrices de las Sociedades Internacionales de Nefrología (ISN) y Nefrología pediátrica (IPNA)10. En una segunda fase, el grupo T-CiS.bcn se propone elaborar un modelo de transición integral del paciente con cistinosis desde los servicios pediátricos a los de adultos, que contemple las necesidades específicas de la enfermedad.
EtiopatogeniaLa cistinosis es una enfermedad hereditaria autosómica recesiva causada por las mutaciones con pérdida de función del gen CTNS (cromosoma 17p13), que codifica la cistinosina11. La cistinosina es una proteína transmembrana específica para el transporte de cistina desde el lisosoma al citoplasma celular12. Su ausencia produce un depósito progresivo de cistina intralisosomal que constituye el principal marcador diagnóstico de la enfermedad1. Se estima una incidencia anual de 1/100.000-200.000 recién nacidos y una prevalencia de 1-9/1.000.000 de población13. La mutación más frecuente en el gen CTNS es una deleción de 57 Kb14, observándose también en población española en un 34% de los pacientes15.
En el interior del lisosoma, el aminoácido cisteína se oxida y forma cistina, compuesto poco soluble. En pacientes con cistinosis se produce un acúmulo de cistina que precipita en forma de cristales en todas las células del organismo, especialmente en tejido renal y ocular16. El aumento de la concentración lisosomal de cistina se asocia al aumento de la apoptosis celular y del estrés oxidativo, y con alteraciones del metabolismo del glutatión y ácido araquidónico17–19. Otros mecanismos patogénicos involucrados son de tipo inflamatorio20 y de sobrecarga o «estrés de retículo endoplásmico», que conducen finalmente a la muerte celular21,22.
ClínicaLa cistinosis es una enfermedad multisistémica23, siendo el riñón y el ojo los primeros órganos afectados. Se han descrito tres formas clínicas: cistinosis nefropática infantil (OMIM#219800) es el subtipo más grave, de aparición precoz; cistinosis nefropática juvenil (OMIM #219900) que es un subtipo menos severo, de debut juvenil o tardío, y de severidad intermedia y la cistinosis no-nefropática o del adulto (OMIM#219750), con afectación ocular exclusiva24. No obstante, en la práctica clínica, se diferencian dos subtipos principales: cistinosis nefropática de debut en la primera infancia con síndrome de Fanconi severo –representa el 95% de todos los casos– y cistinosis no nefropática de debut tardío, que aparece en el joven o adulto, con afectación renal y/o ocular; representa el < 5% del total de afectos. En algunos pacientes la afectación ocular puede preceder en años a la afectación renal25.
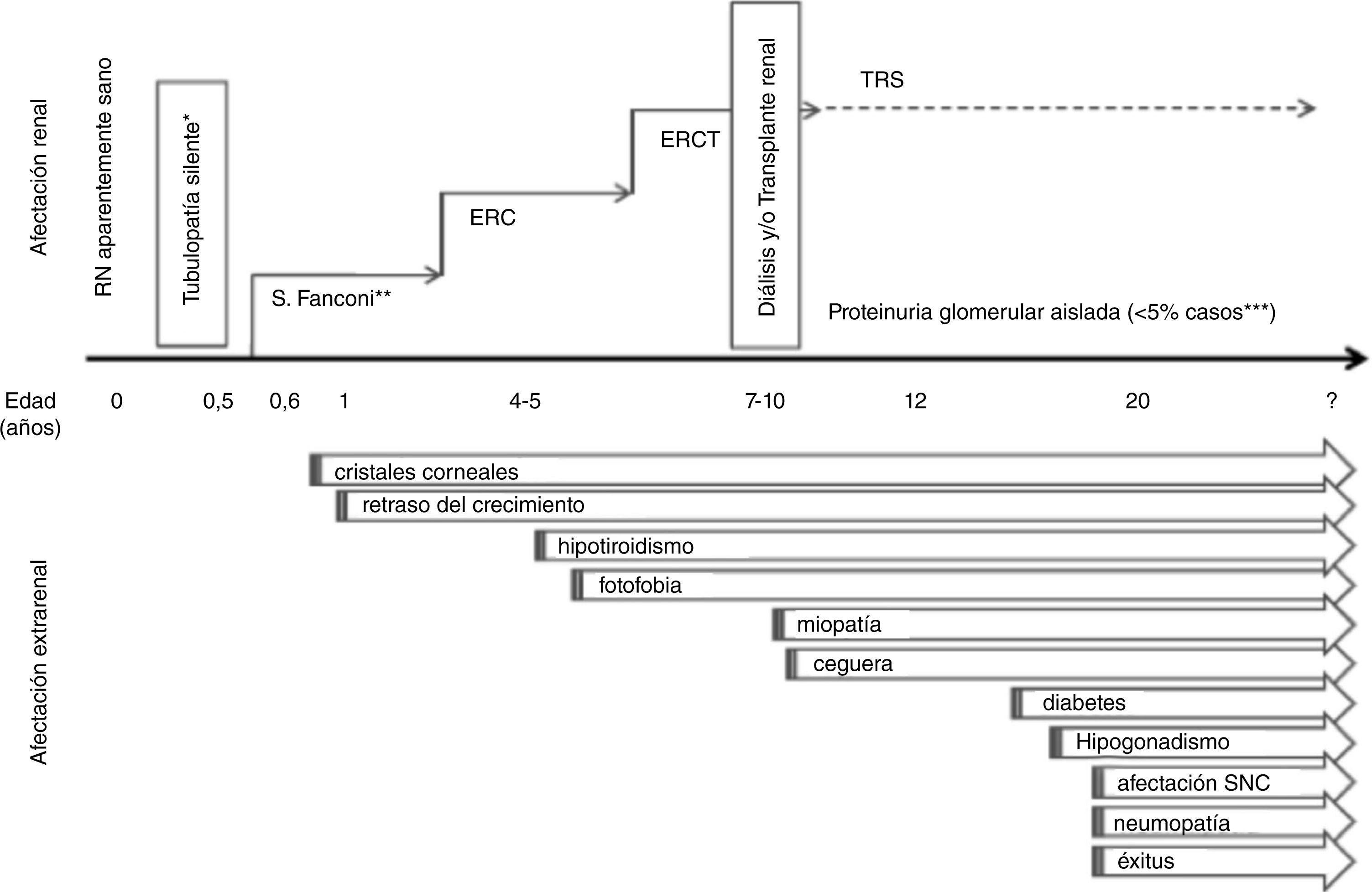
Afectación renalSíndrome de FanconiEl cuadro clínico prototípico consiste en la aparición de un síndrome de Fanconi severo con evolución a la enfermedad renal crónica (ERC) (fig. 1). Es característico que la tubulopatía se manifieste en el segundo semestre de la vida, tras un intervalo libre de síntomas24. Los recién nacidos afectos son aparentemente normales, si bien es posible detectar alteraciones urinarias muy precoces (orina alcalina con glucosuria y/o proteinuria) precediendo a los síntomas26. La cistinosis representa la causa más frecuente de síndrome de Fanconi de etiología genética24 y en lactantes debe ser considerada en primer lugar en el diagnóstico diferencial. No obstante, se han descrito casos de pacientes cistinóticos que debutaron con cuadros atípicos no sugerentes de síndrome de Fanconi sino de tubulopatía distal, tales como diabetes insípida nefrogénica o síndrome de Bartter. De ahí que sea importante considerar el diagnóstico de cistinosis en cualquier lactante con una tubulopatía compleja, sobre todo si presenta afectación del crecimiento y el paciente es anoréxico1. El diagnóstico diferencial debe contemplar la posibilidad de una tubulopatía proximal secundaria27,28. La severidad del síndrome de Fanconi asociado a cistinosis requiere de un riguroso tratamiento con frecuencia muy complejo (tabla 1).
Tratamiento sintomático de la afectación renal de la cistinosis 1,24,29,31
| Objetivo terapéutico | Tratamiento |
| Preservar el balance hídrico con reposición de las pérdidas | Reponer agua según necesidades (entre 1.5 y 6 L/día). Por vía oral o valorar necesidad de sonda nasogástrica o gastrostomía |
| Reducir la poliuria: Indometacina por vía oral (1 a 3 mg/kg/día) | |
| Preservar el balance electrolítico con reposición de las pérdidas | Potasio (entre 2 y 10 mEq/kg/día) |
| ClNa (1-2 mEq/kg/día, con aumento progresivo de la dosis) | |
| Fósforo (entre 1 y 4 g/día) | |
| Neutralización de la acidosis (mantener pH sanguíneo normal y bicarbonato sérico entre 22 y 24 mEq/L) | Bicarbonato o citrato a dosis inicial de 1-2 mEq/kg/día, con aumento progresivo de la dosis) |
| Soporte nutricional | Valoración nutricional con aporte de suplementos calóricos adecuados según la edad y la función renal |
| Tratamiento de la afectación ósea | Colecalciferol |
| Otros | Suplementos de Calcio |
| Vitamina D activa (1Alfacalcidiol, Calcitriol, Paricalcitol, otros) | |
| Hormona de Crecimiento (si está indicado) | |
| Carnitina29 (100 mg/kg/día) | |
| iECAs/ARAs como antiproteinúricos (valorar tolerancia) |
A partir de los dos años de edad, en ausencia de tratamiento específico, se produce una afectación glomerular progresiva con descenso del filtrado glomerular (FG) y aumento de la creatinina plasmática a partir de los 4-6 años, con evolución a ERC avanzada1. Paralelamente, suele atenuarse el síndrome de Fanconi y en consecuencia, es posible reducir los suplementos hidroelectrolíticos (tabla 1). En ausencia del tratamiento farmacológico específico (cisteamina), la edad media de aparición de la enfermedad renal terminal (ERCT) es de 9,2 años. En las series más contemporáneas que incluyen pacientes tratados precozmente con cisteamina, se observa un retraso significativo en la evolución hacia la ERCT alrededor de los 13 años5, hecho que se ha atribuido a un mejor control médico. Además, en los casos con diagnóstico y tratamiento muy precoz, se detecta un porcentaje creciente de pacientes que se mantienen en prediálisis después de la adolescencia6.
Existen formas de cistinosis atenuada o de debut tardío que se manifiestan en la adolescencia o en adultos jóvenes como enfermedad glomerular y proteinuria sin síndrome de Fanconi, aunque ocasionalmente con datos sugestivos de tubulopatía proximal. Habitualmente, los pacientes también presentan manifestaciones oculares de la enfermedad que pueden ser poco sintomáticas25 (fig. 1).
La biopsia renal, aunque no es necesaria para el diagnóstico, demuestra lesiones inespecíficas de glomeruloesclerosis y otras más características como irregularidades en el «borde en cepillo» de la célula tubular proximal, lesiones en «deformidad en cuello de cisne» y ocasionalmente depósitos de cristales de cistina y podocitos gigantes multinucleados2,16,24.
DiálisisEl tratamiento renal sustitutivo (TRS) de elección en la cistinosis es el TxR ya que la enfermedad no recurre en el injerto. No obstante, la limitación de órganos o el diagnóstico tardío condiciona el inicio de diálisis en muchos casos. Según el registro NAPRTCS el 1,4% de los pacientes < 18 años que iniciaron diálisis crónica padecían cistinosis30. Por otro lado, en el registro europeo ESPN/ERA-EDTA Registry un 0,9% de los pacientes < 20 años y un 0,1% de los pacientes > 20 años con TRS padecían cistinosis. En Europa la diálisis peritoneal (DP) representó la modalidad inicial más frecuente (39,6%), seguido por el TxR preventivo (35,1%). Un 17,9% de los pacientes recibió hemodiálisis5 (HD).
El síndrome de Fanconi puede persistir tras el inicio de diálisis, lo que incide en la prescripción dietética del agua y la dieta del paciente y en la necesidad o no de administrar otros fármacos como los quelantes de fósforo. Aunque la pérdida salina urinaria y la poliuria suelen disminuir en la ERC avanzada, es posible que el paciente continúe necesitando suplementos hidroelectrolíticos y carnitina (tabla 1). Raramente la severidad del síndrome de Fanconi justifica realizar nefrectomía de los riñones nativos31. Por otro lado, es característico que muchos pacientes cistinóticos en diálisis presenten afectación extrarrenal que requiere la intervención de otros especialistas de manera integrada (ver apartado de afectación extrarrenal), lo que puede representar un reto para el nefrólogo responsable32.
Trasplante renalComo se ha comentado, el TRS de elección en la cistinosis es el TxR. Las células del injerto no portan el defecto lisosomal y por ello la enfermedad no recurre en el órgano trasplantado. Sin embargo, es posible observar depósitos intersticiales de cristales de cistina, que representan leucocitos del receptor y no tienen significado patológico21. El trasplante de donante emparentado también es curativo y los portadores heterocigotos de la mutación de CTNS pueden ser donantes adecuados ya que no padecen la enfermedad4,6,33.
Existen datos indirectos de pacientes cistinóticos con fallo renal avanzado y de registros internacionales que sugieren la conveniencia de realizar un transplante renal preventivo en esta entidad, sobre todo cuando exista un donante vivo disponible4, evitando el inicio de diálisis5. Por ello la indicación de trasplante renal se establece cuando el FG es < 20ml/min/1,73 m2, algo más precoz que en patología renal de otro origen1.
En EE. UU. (USRDS 2013), la edad media de los pacientes con cistinosis en el primer TxR es de 13,8 años (rango 2-24), cifra que no ha variado en las últimas décadas. De ellos, un 32,4% recibieron un TxR preventivo34. Así mismo, los datos del Registro Europeo (ESPN/ERA-EDTA Registry) muestran un porcentaje similar del 35,1% trasplantados prediálisis, porcentaje mucho más elevado que en otras nefropatías5 (17,1%). Del global de pacientes cistinóticos en TRS, un 85% eran trasplantados renales. Respecto al tipo de donante, en EE. UU., un 54% de los pacientes recibieron un órgano de donante vivo y un 46% de donante cadáver34. De modo similar en Europa el 48,9% recibieron un trasplante de donante vivo5.
Es de destacar que la duración del injerto renal funcionante en los pacientes cistinóticos es superior que el observado en la población trasplantada por otra causa5,35.
Afectación extrarrenalEl mejor pronóstico y el aumento de la supervivencia de los pacientes con cistinosis, ha permitido conocer la afectación multiorgánica de la enfermedad4,6,32,36 (fig. 1).
Afectación ocularLa afectación ocular en la cistinosis es universal. La presencia de cristales de cistina en la córnea es un criterio diagnóstico de esta enfermedad37, si bien su ausencia antes del primer año de vida no excluye que el paciente padezca cistinosis1.
El depósito de cistina en la córnea es una de las manifestaciones más precoces de la cistinosis (fig. 1). Los cristales están ausentes al nacimiento, pero pueden observarse en niños de pocos meses de vida1. Inicialmente se depositan en las capas superficiales de la córnea periférica, pero progresivamente afectan a todas las capas y extensión de la córnea. Sin tratamiento, hay una inexorable progresión del depósito de cristales en la córnea, que aumenta con la edad. La consecuencia del depósito es la fotofobia, que puede ser muy incapacitante, y la alteración en la sensibilidad corneal. Con el tiempo, se producen erosiones corneales recidivantes y edema estromal, que pueden provocar disminución de la agudeza visual. También, se ha descrito depósito de calcio en la membrana de Bowman o queratopatía en banda, que cuando afecta al eje visual provoca disminución de visión37.
Los cristales también se depositan en otras estructuras oculares como la conjuntiva, cámara anterior, iris, cuerpo ciliar, coroides y retina. La afectación retiniana provoca una degeneración de los fotorreceptores, principalmente de los bastones, que altera el campo visual periférico y la visión nocturna, pero que puede reducir la visión central. Más raramente se han descrito sinequias posteriores, adherencia del iris a la cápsula anterior del cristalino y neovascularización de la córnea periférica38,39. Por otro lado, se observa disminución de la producción de lágrimas y ojo seco, y manifestaciones neuro-oftalmológicas (papiledema y oftalmoplejía) secundarias al aumento de la presión intracraneal descrito en esta enfermedad4. En las formas tardías de la enfermedad la presencia de cristales puede no ser detectada hasta la edad adulta25.
Crecimiento y desarrollo. Afectación mineral-óseaEl retraso de crecimiento constituye un síntoma clásico de cistinosis y con frecuencia representa el motivo de consulta precoz36. El mecanismo subyacente es multifactorial, aunque se relaciona con la severidad del síndrome de Fanconi. La concurrencia de acidosis metabólica, hiponutrición, pérdidas digestivas y renales aumentadas y la ERC conducen a un hipocrecimiento que puede ser muy severo40,41. Así mismo, los pacientes presentan alteraciones endocrinas (ver apartado afectación endocrina) y raramente un déficit primario de secreción de hormona de crecimiento42 (GH).
Los pacientes con tratamientos inadecuados son los que presentan una estatura inferior36. Clásicamente la talla adulta descrita en pacientes con tratamiento subóptimo es de 144cm y el peso de 45kg (25cm y 25kg por debajo de la media de la población normal, respectivamente)4. En las series más recientes con mejor control terapéutico se observa menor retraso estatural8 y un impacto favorable del tratamiento sobre los mecanismos reguladores del crecimiento43. No obstante, un 27% de pacientes con cistinosis trasplantados y un 44% en diálisis continúan presentando talla baja en la actualidad5.
La administración precoz de GH mejora la talla, si bien la respuesta terapéutica suele ser inferior a la observada en ERC de otro origen, pese al control óptimo de la enfermedad. La GH constituye una herramienta terapéutica esencial en esta enfermedad, tanto por su efecto sobre el crecimiento en longitud como por su efecto anabólico40,44. En cistinosis se desarrolla una enfermedad ósea metabólica característica causada por distintos factores: el depósito de cristales de cistina en hueso, la mineralización deficiente, el raquitismo de origen renal24 y la ERC per se45. También se han descrito anomalías óseas atribuidas a un déficit de cobre, posiblemente secundario al síndrome de Fanconi46. Por ello es frecuente detectar osteopenia, especialmente en los trasplantados, también en relación con otras alteraciones endocrinas de la enfermedad (ver apartado afectación endocrina) y potencialmente con el tratamiento23,47. En algunos casos se detecta fragilidad ósea y mayor riesgo de fracturas32.
Afectación endocrinaLas manifestaciones endocrinas se producen por destrucción de las glándulas afectas debida a los depósitos de cistina; su incidencia y edad de aparición están asociadas a la instauración del tratamiento específico con cisteamina43.
El hipotiroidismo primario es la complicación endocrinológica más frecuente23, es de curso progresivo y requiere tratamiento crónico con levotiroxina1,4. La diabetes mellitus se caracteriza por una alteración progresiva de la insulinosecreción48, con inmunología negativa y requiere tratamiento con insulina2. Se observa en pacientes trasplantados que reciben corticosteroides23. En los varones se produce hipogonadismo primario y la infertilidad es constante4,49. En las mujeres, sin embargo, ni el hipogonadismo ni la infertilidad son prevalentes, por lo que las pacientes afectas pueden tener hijos50, aunque existe un riesgo aumentado de prematuridad51.
Afectación cardiovascularLa aparición de dislipemia y calcificación vascular por la propia cistinosis y por la ERC per se representan factores de riesgo cardiovascular aumentado23,32,41. Un 42% de los pacientes desarrollan hipertensión arterial habitualmente post-trasplante. También se han descrito aneurismas aórticos y afectación de los vasos coronarios, así como cuadros de cardiomiopatía asociados con el depósito de cristales de cistina en miocardio36. En pacientes adultos se recomienda el despistaje de cardiopatía isquémica4.
Afectación neurológicaLa cistinosis se asocia con alteraciones de la estructura cerebral y aumento de los niveles de cistina en distintas áreas del sistema nervioso y tejido muscular4,6,32,52. En general, las complicaciones neurológicas empeoran el pronóstico de la enfermedad:
- •
Miopatía progresiva isquémica4,32,53 de predominio distal de inicio en las manos; objetivándose además pérdida de masa muscular con posterior afectación de la capacidad ventilatoria y dificultades en la deglución. Algunos autores atribuyen la debilidad muscular que presentan los pacientes al déficit de carnitina24,28,29.
- •
Afectación del Sistema Nervioso Central (SNC)4,31 sobre todo en pacientes con tratamiento subóptimo con cisteamina:
- –
De presentación aguda: epilepsia, ictus, encefalopatía, cefalalgia54–57.
- –
De presentación subaguda/progresiva: hipertensión endocraneal, atrofia cerebral, ataxia, piramidalismo, trastornos en la marcha, calcificaciones en los ganglios basales y periventriculares, desmielinización de la sustancia blanca, deterioro mental58–66.
- •
Alteraciones neurocognitivas67–73: se ha descrito en pacientes cistinóticos un perfil específico de alteraciones en la integración visuomotora, la memoria visual, la atención sostenida, la planificación, la velocidad de procesamiento motor y cálculo aritmético. En consecuencia, presentan una incidencia significativa de dificultades sociales que podría justificar el fenotipo conductual de algunos de los pacientes. La inteligencia suele ser normal.
La detección precoz de complicaciones neurológicas en la cistinosis permite diseñar mejores estrategias terapéuticas, reducir el número de ingresos hospitalarios y mejorar la calidad de vida. La participación de un neurólogo ayudará a evaluar la capacidad funcional de los pacientes, detectando precozmente aquellas manifestaciones neurológicas que puedan afectar la autonomía en las actividades básicas de la vida diaria8,31,32,55.
MisceláneaLa ubicuidad de la cistinosis se pone de manifiesto por sintomatología no específica, como la gastrointestinal, así como otras alteraciones de base genética como la intolerancia al calor y la hipoforesis, entre otras. Así mismo, la naturaleza sistémica de la enfermedad explica la aparición progresiva de otra sintomatología clínica secundaria al depósito de cristales de cistina en los distintos órganos y sistemas, como se detallan a continuación (fig. 1):
- •
Aparato digestivo74:
- o
N usea, vómito, epigastralgia, anorexia
- o
Aumento de la secreción de gastrina (asociada con la toma de cisteamina)
- o
Disminución de la salivación
- o
Dificultad mecánica en la deglución
- o
Retraso de vaciamiento gástrico y dismotilidad intestinal
- o
Pseudo-obstrucción intestinal
- o
Enfermedad inflamatoria intestinal
- •
Hígado32,75:
- o
Hiperplasia nodular regenerativa sin insuficiencia hepática
- o
Hipertensión portal no cirrótica con hiperesplenismo
- o
Colestasis
- o
Hipercolesterolemia
- •
Piel1,76:
- o
Hipopigmentación en piel y cabello por alteración de la melanogénesis
- o
Alteraciones de la sudoración e intolerancia al calor
- •
Médula ósea4:
- o
Anemia
- o
Coagulopatía por plaquetas disfuncionales
El diagnóstico de la cistinosis se establece mediante el diagnóstico clínico y se confirma con el diagnóstico bioquímico y molecular.
Diagnóstico clínicoLos signos guía son el síndrome de Fanconi severo de aparición precoz y la detección de cristales corneales. Evolutivamente es posible observar afectación sistémica (fig. 1). En pacientes con formas menos severas, se observa fallo renal y proteinuria. Ocasionalmente la visualización de cristales en córnea en pacientes adultos con ERC de causa no filiada, conduce al diagnóstico de cistinosis25. La biopsia renal, si bien no es requisito para el diagnóstico de cistinosis, puede ser de utilidad en estas presentaciones atípicas2,16,24,32.
Diagnóstico bioquímico generalSe basa en la detección de trastornos hidroelectrolíticos, del equilibrio ácido-base y eventualmente de la función renal, característicos del síndrome de Fanconi24,28.
Diagnóstico bioquímico específicoConsiste en la detección de niveles elevados de cistina intraleucocitaria en leucocitos totales77. En la actualidad se aplican las técnicas de cromatografía líquida de alta presión-espectrometría de masas en tándem (HPLC-MS/MS) en muestra de granulocitos que es una técnica más sensible78,79. Los valores de referencia son:
- •
individuo sano ≤0,5 nmol 1/2 cistina/mg proteína (los valores > 0,5 tienen significación diagnóstica y se recomienda repetir la determinación)
- •
individuo afecto sin tratamiento >1 nmol 1/2 cistina/mg proteína (habitualmente > 2)
- •
individuo tratado con buen control terapéutico ≤1 nmol 1/2 cistina/mg proteína
Un valor normal de cistina intragranulocitaria en lactantes de corta edad no excluye el diagnóstico de manera absoluta. De ahí que en casos con alta sospecha de cistinosis, se recomiende realizar una segunda determinación a los 3-6 meses de la primera, cuando esta no sea concluyente79 (tabla 2).
Diagnóstico bioquímico y monitorización de niveles de cistina. Diagnóstico molecular. (www.orpha.net)
| Requerimientos para la determinación de cistina intragranulocitaria77–79 | |
| Condiciones de obtención de muestra | No precisa ayuno |
| En pacientes tratados, la extracción debe realizarse a las 6 horas post-dosis cisteamina | |
| Tipo de tubo | Con heparina de litio o sódica. |
| Volumen mínimo | Pacientes con <10 kg: 6mL de sangre total |
| Pacientes con ≥ a 10 kg: 10mL de sangre total | |
| Condiciones de conservación y envío | Una vez obtenida la muestra, enviarla inmediatamente a Tª ambiente, ya que debe llegar al laboratorio antes de 24 horas. |
| RECOMENDACIONES para la monitorización de los niveles de cistina | Al inicio del tratamiento y mensualmente hasta alcanzar niveles adecuados por ajustes de dosis de cisteamina |
| En pacientes estables: cada 6 meses | |
| Se aumentará la frecuencia de monitorización en aquellas situaciones de cambios clínicos significativos (TxR y D) | |
| LABORATORIO de Referencia en España: Dirección y contacto | |
| Hospital Clínic de Barcelona | |
| Servicio de Bioquímica y Genética Molecular. Sección de Errores Congénitos del Metabolismo | |
| Dra. Judit García Villoria: jugarcia@clinic.ub.es | |
| Tfn. 93 227 56 00 Ext. 7585 | |
| C/ Mejía Lequerica s/n. Edificio Helios III. Planta baja. 08028, Barcelona. | |
| Requerimientos para el diagnóstico molecular del gen CTNS12,13,15,80 | |
| Estudio mutacional de pacientes y familiares | |
| Muestra | 2-3mL de sangre en EDTA a Tª ambiente |
| DNA a Tª ambiente | |
| Diagnóstico prenatal | |
| Preferentemente biopsia corial o en amniocitos cultivados. | |
| Se requiere la detección previa de mutación en el caso índice y en progenitores | |
| Es indispensable concertación previa con el laboratorio. | |
| LABORATORIO de Referencia en España: Dirección y contacto | |
| Hospital Clínic de Barcelona | |
| Servicio de Bioquímica y Genética Molecular. Sección de Errores Congénitos del Metabolismo | |
| Dra. Mª Josep Coll: mjcoll@clinic.ub.es | |
| Tfn. 93 227 9341 | |
| C/ Mejía Lequerica s/n. Edificio Helios III. Planta baja. 08028, Barcelona. | |
La cistinosis se confirma con la detección de mutaciones en homocigosis o heterocigosis compuesta en el gen CTNS. Se han descrito > 100 mutaciones diferentes y la más frecuente es la deleción ∼57kb de los primeros 10 exones, sobre todo en pacientes con ascendente norte-europeo. Las mutaciones puntuales se traducen en la ausencia de proteína o en una proteína truncada probablemente no funcional11,12,14,15,80 (tabla 2).
Consejo genéticoAl tratarse de una enfermedad autosómica recesiva, la probabilidad de una familia con un hijo afecto, de tener un segundo hijo con cistinosis, es del 25%81. El consejo genético incluye, en este caso, la información sobre las técnicas de diagnóstico prenatal y selección de embriones82,83. La probabilidad que tiene una mujer con cistinosis de tener un hijo afecto es muy bajo excepto en familias consanguíneas o poblaciones endogámicas. Los varones con cistinosis son universalmente estériles49.
El consejo genético suele incluir información referente a las asociaciones de pacientes84–86 y las estrategias institucionales en enfermedades minoritarias87.
TratamientoTratamiento sintomático de la afectación renalLos objetivos del tratamiento incluyen el control del síndrome de Fanconi (tabla 1) y sus complicaciones (Anexo 1) y el de los factores involucrados en la progresión del fallo renal24–28. En ERCT es prioritario promover el TxR. Independientemente de la afectación renal, todos los pacientes deberán recibir tratamiento específico con cisteamina para la prevención y el control terapéutico de la enfermedad multisistémica.
El tratamiento de la ERC seguirá las guías internacionales88–90. En los pacientes trasplantados se recomienda minimizar o evitar los corticoides23.
Tratamiento específico con cisteaminaCisteamina oralEl tratamiento específico de todas las formas clínicas de cistinosis es la cisteamina oral. La cisteamina disminuye el contenido intralisosomal de cistina formando complejos disulfuro cisteína-cisteamina capaces de salir de los lisosomas mediante un canal alternativo a la cistinosina, que es el transportador de lisina3,19,91.
El primer tratamiento farmacológico específico para la cistinosis es Cystagon® (bitartrato de cisteamina oral en cápsulas duras) siendo el único autorizado en España92–94. Recientemente ha sido aprobada una nueva fórmula de cisteamina en cápsulas duras gastrorresistentes93–96.
Beneficios terapéuticosLa cisteamina oral debe introducirse desde el momento del diagnóstico y mantenerse durante toda la vida. Cuando la adherencia es consistente, la cisteamina es capaz de deplecionar hasta un 95% los depósitos celulares de cistina62. La reducción de estos depósitos se correlaciona con la gravedad de la cistinosis32. Se ha demostrado que la cisteamina prolonga la vida del paciente, retrasa la progresión de la enfermedad renal y el inicio de tratamiento renal sustitutivo5,97. Así mismo, disminuye la severidad y frecuencia de las manifestaciones extra-renales32. El pronóstico de la enfermedad está directamente relacionado con el inicio precoz y la duración del tratamiento. Incluso cuando el diagnóstico de cistinosis es tardío, la administración de cisteamina ha demostrado beneficios clínicos4,6,98. Aunque el síndrome de Fanconi no suele ser reversible con cisteamina19, en algunos casos aislados de diagnóstico prenatal, la instauración de cisteamina en las primeras semanas de vida evitó la aparición de tubulopatía99,100.
Cystagon®: posología, administración y monitorización del tratamientoLa base del tratamiento consiste en la depleción de cistina intralisosomal que en la práctica clínica se refleja en la reducción de los niveles de cistina intragranulocitaria, con un objetivo terapéutico óptimo fijado por debajo de 1 nmol hemicistina/mg de proteína. La disminución de estos niveles se correlaciona con la concentración plasmática de cisteamina durante las seis horas siguientes a la administración de Cystagon®, siendo mínimos a ∼2 horas de la toma del fármaco y retornando a su valor basal (pre-dosis) 6 horas después. Esto explica la necesidad de administrar el fármaco cada 6 horas, toma nocturna incluida101,102 (tabla 3).
Tratamiento específico con cisteamina
| Cisteamina oral - Cystagon®31,47,92–94,102 | ||
|---|---|---|
| Posología | ||
| Según edad | Niños ≤12 años | Pacientes >12 años |
| Dosis recomendada | en función del área de superficie corporal (g/m2/día) | en función del peso cuando >50kg |
| 1.30 g/m2/día dividida en cuatro tomas al día | 2 g/día dividida en cuatro tomas al día | |
| Dosis inicial | Para evitar intolerancias, administrar de 1/4 a 1/6 de la dosis de mantenimiento prevista y aumentar progresivamente durante un periodo de tiempo entre 4 y 6 semanas | |
| Ajustes dosis | La dosis debe aumentarse si la tolerancia es adecuada y si el nivel de cistina intragranulocitaria es superior a: | |
| >1 nmol hemicistina/mg proteína | ||
| Dosis máxima | 1.95 g/m2/día | |
| La sobredosificación no está aconsejada por no mejorar el pronóstico y asociarse con efectos adversos47 | ||
| Insuficiencia renal | No precisa modificación de la dosis | |
| Administración | 4 tomas al día; cada 6 horas. Toma nocturna incluida | |
| En menores de 6 años se recomienda abrir la cápsula y esparcir su contenido sobre los alimentos | ||
| Evitar administrar con bebidas ácidas | ||
| Ajustar la dosis en función de los niveles de cistina intragranulocitaria | ||
| Reacciones adversas | Trastornos gastrointestinales por hipersecreción ácida | |
| Recomendaciones para mejorar la tolerabilidad digestiva: | ||
| Administración concomitante de inhibidores de la bomba de protones102 | ||
| Administrar en las comidas o inmediatamente después. Se recomienda ingesta simultánea con alimentos como la leche, las patatas y otros alimentos ricos en almidón92 | ||
| Olor corporal específico y halitosis | ||
| Recomendaciones para mejorar olor: puede aminorarse con pastillas mentoladas31 | ||
| Otras reacciones adversas: consultar ficha técnica92 | ||
| Interacciones medicamentosas | No se han realizado estudios de interacciones. | |
| Se puede administrar conjuntamente con suplementos electrolíticos o minerales, análogos de la vitamina D, tiroxina o inmunosupresores | ||
| Cisteamina tópica oftálmica36–38,98,99 | ||
| Tratamiento oftalmológico tópico | Solución salina de cisteamina al 0.55% (Anexo 2) | |
| Posología | 1 gota en cada ojo | |
| Administración | 10-12 veces al día | |
Se recomienda monitorizar los niveles al inicio del tratamiento y mensualmente tras los cambios en la dosis prescrita. En pacientes en seguimiento con niveles estables, se recomienda la monitorización cada 6 meses. Así mismo, de modo individualizado, se aumentará la frecuencia de monitorización en aquellas situaciones de cambios clínicos significativos77–79 (TxR y D) (tabla 2).
Tratamiento oral en situaciones especialesEnfermedad renal crónica, diálisis y trasplanteNo existe una correlación entre el FG y los niveles de cisteamina en plasma, por lo que no se precisa ajuste de dosis por función renal, sino que la dosis prescrita del fármaco se debe ajustar a la cuantificación de los niveles de cistina intragranulocitaria. Tampoco se precisa ajustes por síndrome de Fanconi6,103.
Embarazo y lactanciaAunque no existen datos suficientes se ha observado en animales toxicidad reproductiva y efectos teratogénicos de la cisteamina104. Por ello está contraindicada durante el embarazo, especialmente durante el primer trimestre. Se recomienda la planificación familiar en mujeres en edad reproductiva. Además, debe evitarse su administración durante la lactancia.
Cisteamina tópica (gotas oftálmicas)El tratamiento específico de la afectación ocular de la cistinosis requiere, además de la cisteamina oral, la administración de cisteamina tópica en forma de gotas oftalmológicas. La estrategia terapéutica oftalmológica32,37 distingue entre:
Afectación de la córneaLos depósitos de cristales de cistina deben tratarse con la administración tópica de cisteamina dado que la córnea es una estructura avascular y como consecuencia el fármaco oral no es eficaz a nivel de córnea37,38,105,106. La prescripción recomendada se indica en la tabla 3. Actualmente se están desarrollando formulaciones viscosas, para conseguir una mayor permanencia de la cisteamina en contacto con la superficie ocular y poder disminuir la frecuencia de instilación con igual eficacia107,108.
Afectación de estructuras no cornealesLa cisteamina oral es eficaz a nivel de la retina y otras estructuras oculares. La incidencia de retinopatía ha disminuido por el uso sistémico de cisteamina. La frecuencia y gravedad de las manifestaciones oftalmológicas no corneales están directamente relacionadas con la adherencia al tratamiento oral con cisteamina, con riesgo de pérdida importante de la visión si no se realiza un tratamiento sistémico correcto39,109.
Adherencia al tratamiento específico con cisteaminaLa Organización Mundial de la Salud (OMS) define la adherencia como el grado en que el comportamiento de una persona se corresponde con las recomendaciones acordadas con los profesionales sanitarios110.
El impacto de la no-adherencia en cistinosis se refleja en un peor pronóstico y una mayor progresión de la enfermedad renal y extrarrenal en los pacientes no adherentes respecto a los cumplidores5,6,97
La información sobre adherencia en pacientes con cistinosis es limitada, si bien la monitorización de niveles de cistina intragranulocitaria permite detectar a los pacientes no cumplidores77,79. Otros estudios han constatado una adecuada adherencia a Cystagon® en el paciente pediátrico que disminuye significativamente en adolescentes y adultos7,8. No obstante, en grupos de pacientes muy motivados, se ha descrito que únicamente un 8% presenta problemas de adherencia111.
En cistinosis concurren factores de riesgo de no adherencia a cisteamina tales como: la pauta posológica, los problemas de tolerancia, efectos secundarios y los requerimientos de múltiples medicaciones para el control de las manifestaciones clínicas de la enfermedad. Otros factores de riesgo no menos importantes y no exclusivos de cistinosis son: el conocimiento limitado sobre la enfermedad, la falta de motivación, la transición inadecuada del paciente a los servicios de adultos y el impacto de la enfermedad en la calidad de vida7,9,10.
No obstante, la adherencia subóptima al tratamiento no es un fenómeno restringido a la cistinosis. Estudios recientes describen que el 52-67% de los pacientes adultos trasplantados renales, no siguen correctamente incluso la pauta inmunosupresora prescrita, aumentando la probabilidad de pérdida de injerto112,113. Estos porcentajes son similares a los publicados en pacientes con cistinosis en nuestra población7, lo que puede indicar la coexistencia de escenarios comunes a la ERC. Por lo tanto, en aras a mejorar la adherencia en pacientes cistinóticos, se recomiendan estrategias correctoras de los factores de riesgo a la no-adherencia y de promoción del autocuidado del paciente, similares a las implementadas con éxito en adultos con trasplante renal114–119 (tabla 4).
Recomendaciones para mejorar la adherencia al tratamiento7,10,114–119
| Identificar factores de riesgo que afectan a la adherencia y aplicar medidas correctivas en lo posible: |
| Factores intrínsecos al paciente y socioeconómicos |
| Factores relacionados con la enfermedad |
| Factores relacionados con el tratamiento |
| Barreras de organización sanitaria |
| Identificar y asignar el “coordinador/gestor del paciente” Promover la educación del paciente y el soporte al tratamiento: |
| Implementar programas de educación en la enfermedad |
| Acordar planes terapéuticos: de fácil seguimiento y con medidas de soporte para el cumplimiento terapéutico |
| Utilizar cuestionarios para la detección de la no adherencia |
| Seguimiento de las citas médicas y las ausencias |
| Desarrollar programas de soporte al paciente, involucrando familiares, amigos y asociación de pacientes. |
| Crear un equipo médico multidisciplinar |
| Implementar programas de transición protocolizados a la medicina del adulto |
El grupo T-CiS.bcn, tras una revisión exhaustiva de la literatura médica en cistinosis y analizada la experiencia clínica con nuestros pacientes, hemos elaborado las siguientes recomendaciones de atención integral de la enfermedad y la transición del paciente adolescente. Nuestro objetivo es proporcionar una herramienta de soporte y asesoramiento clínico a otros profesionales de la salud involucrados en el cuidado de los pacientes con cistinosis.
Estas recomendaciones se presentan a continuación en las tablas 4–8.
Recomendaciones para el seguimiento del paciente en tratamiento renal sustitutivo: diálisis y trasplante
| Recomendaciones en Diálisis6,103 |
| Promover el TxR preventivo como modalidad inicial de tratamiento renal sustitutivo en pacientes con ERC avanzada |
| Monitorizar el volumen de diuresis residual y la pérdida salina urinaria para adaptar la prescripción de diálisis y evitar la ultrafiltración excesiva |
| Mantener el tratamiento general del Síndrome de Fanconi y adaptar la dieta de modo individualizado |
| Monitorizada cuidadosamente la afectación extrarenal con un enfoque multidisciplinar de todos los especialistas implicados |
| Es necesario mantener el tratamiento oral y tópico ocular con cisteamina |
| No se debe ajustar la dosis de cisteamina al filtrado glomerular (Ver apartado Tratamiento específico con cisteamina) |
| Recomendaciones en Trasplante renal4–6,33,88–90 |
| Entrada en lista de espera |
| Impulsar la realización de trasplante renal preventivo cuando el filtrado glomerular ≤20 ml/min/1.73 m2 |
| Donante vivo o cadáver |
| Evaluación del síndrome de Fanconi asociado (diuresis residual –puede ser muy elevada–, pérdida salina, raquitismo, acidosis tubular, déficit de carnitina). |
| Monitorizar niveles de cistina intragranulocitaria y optimizar el tratamiento con cisteamina (oral y tópica) |
| Valoración de la posible afectación sistémica y su impacto sobre el trasplante (hipotiroidismo, diabetes, enfermedad cardiovascular, enfermedad ósea, trastornos deglutorios) |
| Prescripción de líquidos y dieta individualizada. Valoración de necesidades de suplementos de fosfato, potasio, bicarbonato y carnitina |
| Pre-trasplante y trasplante inmediato |
| Evitar depleción de volumen en el preoperatorio y durante la cirugía (fluidoterapia endovenosa intensiva para garantizar la euvolemia, incluyendo suplementos de K y Bicarbonato) |
| Inmunosupresión según protocolo del centro |
| Suspensión transitoria del tratamiento con cisteamina |
| Post-trasplante inmediato |
| Fluidoterapia y administración electrolítica suficiente para mantener un balance hidroelectrolítico adecuado y un buen control del síndrome de Fanconi residual |
| Vigilar la posible aparición de diabetes |
| Inmunosupresión según protocolo del centro |
| Atención continuada post-trasplante |
| Reintroducir la cisteamina una vez que el paciente y el trasplante se encuentren estables aproximadamente a las 3-4 semanas post-trasplante en dosis crecientes hasta la dosis terapéutica |
| Monitorizar los niveles de cistina intragranulocitaria |
| Inmunosupresión según protocolo del centro, promoviendo reducción y/o suspensión de corticoides |
| Controles y seguimiento de acuerdo a las recomendaciones y guías clínicas |
| Mantener el tratamiento tópico ocular con cisteamina y estimular el cumplimiento terapéutico |
| Valoración de la posible afectación sistémica y su impacto sobre el trasplante. Promover y estandarizar un plan de atención que incluya el cuidado multidisciplinar de la cistinosis |
En cualquier situación clínica, se precisa tratamiento con cisteamina oral para mantener los niveles de cistina recomendados <1 nmol hemicistina/mg proteína y cisteamina tópica para eliminar los depósitos corneales
Recomendaciones para el seguimiento oftalmológico4,37–39,105–109,120
| Tipo de exploración | Estructura ocular | Frecuencia | Observaciones |
|---|---|---|---|
| Biomicroscopía con lámpara de hendidura | Estudio córnea y resto segmento anterior | Anual | |
| Medir presión intraocular | Descartar hipertensión ocular | Anual | |
| Fondo de ojo bajo midriasis | Valorar papila y pigmentación de la retina | Anual (En pacientes con GH valoración basal y a los 4 meses de rutina para detectar hipertensión intracraneal120) | Urgente: si el paciente refiere disminución severa AV (infrecuente) |
| ERG fotópico y escotópico | Funcionalidad conos y bastones | Solo si el paciente refiere alteración visión nocturna o alterado el fondo de ojo | |
| Instrumental necesario | Lámpara de hendidura* | ||
| Tonómetro | |||
| Oftalmoscopio indirecto |
* Muy sensible para diagnosticar los cristales de cistina en la córnea pero es menos útil para el seguimiento dado que la cuantificación de los cristales es bastante subjetiva. Por ello es interesante recoger en cada exploración, de forma detallada la distribución de los cristales en la córnea, especificando si solo se depositan en la periferia o de forma difusa y si se localizan en epitelio, estroma y/o endotelio.
Subrayar la importancia del tratamiento con cisteamina oral y cisteamina tópica
ERG: electroretinograma; GH:hormona de crecimiento; AV: agudeza visual.
Recomendaciones para el seguimiento y tratamiento de la afectación endocrina4,5,32,48,49,121
| Trastorno | Exámenes complementarios | Frecuencia | Tratamiento | Observaciones |
|---|---|---|---|---|
| Hipotiroidismo | TSH, T4L, Ac anti-tiroideos. No es necesaria prueba de imagen | Al diagnóstico | Tiroxina | Iniciar tratamiento cuando TSH > 10 mUI/L |
| TSH, T4L | Control rutinario: Trimestral (ajuste dosis) | Si hay síntomas valorar inicio con TSH entre 5-10 mUI/L | ||
| Anual, si TSH normal | ||||
| Diabetes Mellitus | Glucemia, HbA1c (opcional: c-péptido). | Diagnóstico | Insulina | Valorar inicialmente dosis única diaria de insulina retardada, si reserva pancreática suficiente. |
| Si síntomas poliuria-polidipsia: bioquímica con ionograma, Equilibrio venoso y cetonurias | ||||
| Glucemia, HbA1c | Control trimestral o bianual, según criterio clínico. | |||
| Anual, en ausencia de clínica. | ||||
| Perfil lipídico: colesterol total, LDL, HDL y TG | Anual | Tratamiento dislipemia | ||
| Fondo de ojo | Según criterio oftalmólogo | |||
| Sensibilidad algésica y vibratoria | Educación para la | |||
| Exploración pie y pulsos | prevención lesiones | |||
| Retraso Pondoestatural | Valoración nutricional | Según criterio clínico | Optimizar nutrición | Valorar suplementos y consulta a dietética |
| Peso y Talla | En cada visita | Hasta completar la maduración ósea | ||
| Ver apartado S.Fanconi | Tratamiento del S.Fanconi | |||
| Edad ósea GH, IGF-1, IGFBP-3 Si no insuficiencia renal: pruebas de secreción de GH | Diagnóstico | r-GH | Descartar hipogonadismo en el paciente puberal. En paciente trasplantado valorar retirada o disminución de | |
| Edad ósea, IGF-1 | Control anual | cortidoides | ||
| Examen de fondo de ojo | Antes de iniciar GH y a los 3-4 meses de tratamiento | La hipertensión intracraneal secundaria a GH suele presentarse al inicio del tratamiento (media 3-4 meses). | ||
| Urgente si cefalea o disminución de la visión. | ||||
| Calcio, fósforo, fosfatasa alcalina, 25OHD3, PTH | Anual | Vitamina D, si déficit.Calcitriol, si insuficiencia renal. | Descartar raquitismo y déficit nutricional de vitamina D | |
| Densitometría ósea | En la edad adulta. Control a valorar según resultado. | Si osteo-porosis, valorar tratamiento específico | ||
| Hipogonadismo | Exploración física: maduración sexual y caracteres sexuales secundarios | En cada visita | Periodo puberal hasta la maduración completa | |
| Testosterona, SHBG, LH, FSH + valoración de función tiroidea (ver más arriba) | Diagnóstico | Testosterona transdérmica diaria (si pánico a inyecciones), o Testosterona IM depot (si problemas de cumplimiento) | Si testosterona baja, con LH y FSH normales: Resonancia magnética hipofisaria | |
| T, LH, FSH | Control anual | |||
| Fertilidad | Seminograma | Diagnóstico | La infertilidad es una característica constante en los varones afectos | |
| Si embarazo, considerar de alto riesgo | Suspender cisteamina oral durante gestación | |||
En cualquier situación clínica, se precisa tratamiento con cisteamina oral para mantener los niveles de cistina recomendados <1nmol hemicistina/mg proteína y cisteamina tópica para eliminar los depósitos corneales.
25OHD3: 25-hidroxicolecalciferol (calcifediol); FSH: Folículoestimulina; GH: Hormona del crecimiento; HbA1c: Hemoglobina glicosilada; IGF-1: Factor de crecimiento 1 similar a la insulina; IGFBP3: proteína transportadora 3 del factor de crecimiento similar a la insulina; LH: Luteotropina; PTH: Parathormona; rGH: Hormona del crecimiento recombinante.; SHBG: Globulina fijadora de hormonas sexuales; T: testosterona; T4L: Tiroxina libre; TG: Triglicéridos; TSH: Tirotropina
Recomendaciones para el seguimiento y tratamiento de la afectación neurológica4,52–73,122–126
| Trastorno | Evaluación | Exámenes complementarios | Frecuencia | Tratamiento | Observaciones |
|---|---|---|---|---|---|
| Función motora de la musculatura esquelética 4,52,53 | Grado de atrofia muscular Progresión de la enfermedad Grado de discapacidad | Escala MRC | Anual | Rehabilitación | Utilizar instrumentos validados |
| Determinaciones cuantitativas de la fuerza muscular en las manos (dinamómetro de Jamar, vigorímetro de Martin y Jamar Hidraulic Pinch Gauge) | Anual | ||||
| Electromiograma | Según criterio clínico | ||||
| RM muscular | Según criterio clínico | ||||
| Biopsia muscular | Según criterio clínico | ||||
| Función motora oro-facial (lenguaje) y deglutoria57,122,123 | Musculatura facio-bulbar: fuerza y rango del movimiento de labios, lengua, velo del paladar, mandíbula y músculos faciales en la fonación, la articulación, la deglución, la respiración y la expresión | Exploración física dirigida | Anual | Reeducación si se detecten los primeros síntomas de disfagia con el fin de prevenir broncoaspirados | |
| Videofluoroscopia | Según criterio clínico | Objetivo: evaluar las diferentes fases de la deglución y el movimiento de un bolo de comida dentro de la cavidad oral y su paso a través del esófago | |||
| Función muscular respiratoria 124 | Presencia de disnea, apneas y/o ronquidos durante el sueño, cefaleas matutinas, hipersomnia diurna, pérdida de fuerza en la tos o aumento de anómalo en la expectoración | Espirometria | Anual | Sintomático Fisioterapia respiratoria, soporte ventilatorio (CPAP). | Especialmente en pacientes con trastornos de deglución, riesgo de broncoaspiración o que presenten síntomas de insuficiencia respiratoria neuromuscular |
| Saturación de oxígeno Gasometría Polisomnografía | |||||
| Sistema Nervioso Central 54,59–68,125,126 | Signos guía de afectación (cefalea, episodios de pérdida de conocimiento, rendimiento escolar defectuoso, deterioro en funciones cognitivas, alteraciones en la conducta, etc.) | Exploración física dirigida | Anual | Dirigido al trastorno subyacente | Pruebas complementarias si se detectan anomalías. |
| Pruebas neurofisiológicas | Según criterio clínico | ||||
| Neuroimagen | Según criterio clínico | ||||
| Alteraciones neurocognitivas 69–73 | Exploración neuropsicológica | Cuestionarios específicos evaluadores del rendimiento cognitivo global, la atención y las funciones ejecutivas, el lenguaje, la memoria, las funciones perceptivas, visuoespaciales y visuoconstructivas, así como el control motor cognitivo voluntario | Anual | Dirigido al trastorno subyacente | Adaptados a la edad del paciente (niños en etapa escolar y adultos) |
Los autores declaran no tener ningún conflicto de interés.
| Principio activo | Especialidad farmacéutica/Producto | Composición | Categoría |
|---|---|---|---|
| Suplementos alcalinos | |||
| Sódio bicarbonato | Bicarbonato de sosa Torres Muñoz 500mg (30 comp.) | 500 mg/comp. | EFP |
| Bicarbonato de sosa Torres Muñoz 60g; 200g; 750g (polvo) | EFP | ||
| Bicarbonato sod Serra 180g (polvo) | |||
| Bicarbonato sod Viviar 210g; 250g; 500g (polvo) | EFP | ||
| Bicarbonato sódico NM 1g; 2g (sobres) | 1 g/sobre; 2 g/sobre | PF | |
| Bicarbonato sódico 1M (8,4%) solución oral | 1 mEq/ml | FM | |
| Citrato sódico | Bicitra solución oral | 1 mEq Bicarbonato/ml; 1 mEq Na/ml; 0,5 mmol citrato/ml | FM |
| Citrato potásico | Polycitra solución oral | 2 mEq Bicarbonato; 1 mEq Na/ml; 1 mEq K/ml; 1 mmol Citrato/ml | FM |
| Polycitra LC con fósforo solución oral | 2 mEq Bicarbonato; 1 mEq Na/ml; 1 mEq K/ml; 1 mmol Citrato/ml; 0,6 mmol P/ml) | FM | |
| Suplementos de potasio | |||
| Potasio ascorbato | Boi-K (20 comp. efervescentes) | 10 mEq K/comp. | EF |
| Bok-K Aspártico (20 comp. efervescentes) | 25 mEq K/comp. | EF | |
| Potasio cloruro | Potasion 600mg (60 cápsulas) | 8 mEq K/cápsula | EF |
| Potasio glucoheptonato | Potasion 1,32g/5ml (125ml; 250ml) | 1 mEq K/ml | EF |
| Suplementos de fósforo | |||
| Fosfato sódico | Solución de fósforo oral o solución de Joulie | 1 mmol P/ml (30,9mg P/ml) | FM |
| Phosphate sandoz 500mg (100 comp) | 16,1 mmol P/comp (500mg P/comp.) | EX | |
| Fosfato sódico monobásico NM (100 sobres) | 26 mmol P/sobre (800mg P/sobre) | PF | |
| Polycitra LC con fósforo solución oral | 2 mEq Bicarbonato; 1 mEq Na/ml; 1 mEq K/ml; 1 mmol Citrato/ml; 0,6 mmol P/ml) | FM | |
| Otros | |||
| Carnitina | Carnicor 1,5g/5ml (40ml solución oral) | 300 mg/ml carnitina | EF |
| Carnicor 1g viales bebibles (10ml) | 100 mg/ml carnitina | EF | |
| Secabiol 300mg/ml (40ml solución oral) | 300 mg/ml carnitina | EF | |
| Indometacina | Artinovo 25mg (30 cápsulas) | 25mg indometacina | EF |
| Flogoter 25mg (40 cápsulas) | 25mg indometacina | EF | |
| Inacid 25mg (30 cápsulas) | 25mg indometacina | EF | |
| Indonilo 25mg (24 cápsulas) | 25mg indometacina | EF | |
| Indometacina 2 mg/ml solución oral | 2 mg/ml indometacina | FM | |
| Cisteamina | Cisteamina 0,55% colirio | 0,55% cisteamina | FM |
Comp: comprimido; PF: producto parafarmacia; EFP: especialidad farmacéutica publicitaria; FM: fórmula magistral; EF: especialidad farmacéutica financiable; EX: especialidad farmacéutica extranjera
| Fórmulas magistrales líquidas | ||
|---|---|---|
| Fórmula magistral | Composición | Cantidad |
| Bicarbonato sódico 1M (8,4%) solución orala | Bicarbonato sódico | 8,4 g |
| Agua destilada estéril (csp) | 100 ml | |
| Bicitra solución oralb | Sodio, citrato 2H2O (Tri-) | 10 g |
| Ácido cítrico monohidratado | 6,7 g | |
| Jarabe simple con conservantes | 50 ml | |
| Agua destilada estéril | 40 ml | |
| Polycitra solución oralb | Potasio, citrato H2O (Tri-) | 11 g |
| Sodio, citrato 2H2O (Tri-) | 10 g | |
| Ácido cítrico monohidrato | 6,7 g | |
| Jarabe simple con conservantes | 50 ml | |
| Agua destilada estéril | 38 ml | |
| Polycitra LC con fósforo solución oralc | Sodio hidrogenofosfato-12 | 1,4 g |
| Ácido fosfórico 85% Ph.Eur | 1,4 ml | |
| Aigua destilada estéril | 32 ml | |
| Potasio, citrato H2O | 11 g | |
| Sodio, citrato | 10 g | |
| Ácido cítrico monohidrato | 6,7 g | |
| Jarabe simple (csp) | 100 ml | |
| Esencia de naranjas dulces | 1 gts | |
| Indometacina 2 mg/ml solución orald,e,f,g | Indometacina | 0,2 g |
| Alcohol etílico | 0,7 ml | |
| Agua destilada estéril | 0,3 ml | |
| Jarabe simple+ Nipagin/nipasol | 100 ml | |
| Fosfatos solución oral (Solución de Joulie)b | Ácido orto-fosfórico 85% | 5,45 g |
| Fosfato disódico 12 H2O (di) | 18,72 g | |
| Agua destilada estéril (csp) | 100 ml | |
| Cisteamina 0,55% colirioh | Benzalconio cloruro | 0,045 g |
| Sodio cloruro 0,9% | 225 ml | |
| Cisteamina clorhidrato | 1,2375 g | |
Trissel LA. Trissel's Stability of Compounded Formulations. 3rd Ed. American Pharmacists Association, Washinton DC; 2005:388-389
The United States Pharmacopeial convention. USP-Pharmacists’ Pharmacopeia, 2nd Ed., Rockville MD; 2008



