













Cystinosis is a rare systemic lysosomal storage disease that mainly affects the kidney and the eye. Renal replacement therapy is started in patients with cystinosis during the first decade of life in the absence of treatment. The prognosis of cystinosis depends on early diagnosis and the prompt start of and good compliance with cysteamine treatment. Kidney disease progression, extra-renal complications and shorter life expectancy are more pronounced in patients who do not adhere to treatment.
ObjectiveThe aim of this work was to establish recommendations for the comprehensive care of cystinosis and facilitate patient transition from paediatric to adult medicine, based on clinical experience. The goal is to reduce the impact of the disease and improve prognosis and patient quality of life.
MethodsBibliographic research and consensus meetings with a multidisciplinary professional team of clinical experts in cystinosis (T-CiS.bcn group) from 5 hospitals in Barcelona.
ResultsThis consensus document gathers specific recommendations for the diagnosis, treatment and multidisciplinary care of cystinotic patients in the following areas: nephrology, dialysis, kidney transplantation, ophthalmology, endocrinology, neurology, laboratory, genetic counselling, nursing and pharmacy.
ConclusionsGuidelines for the comprehensive care of cystinosis provide a support tool for health professionals who look after these patients. They are based on the following main pillars: (a) a multidisciplinary approach; (b) appropriate disease monitoring and control of white blood cell (WBC) cystine levels; (c) the importance of adherence to cysteamine treatment; and (d) the promotion of patient self-care by means of disease education programmes. All these recommendations will lead us, in a second phase, to create a coordinated model of transition from paediatric to adult care services which will cover the specific needs of cystinosis.
La cistinosis es una enfermedad lisosomal minoritaria de expresión sistémica con especial afectación renal y oftalmológica, en la que los pacientes inician terapia renal sustitutiva en la primera década de la vida en ausencia de tratamiento. El pronóstico de la cistinosis depende del diagnóstico precoz, la pronta instauración del tratamiento con cisteamina y el buen cumplimiento terapéutico. La progresión de la enfermedad renal y de las complicaciones extrarrenales y una menor supervivencia, son más acentuadas en pacientes no adherentes.
ObjetivoEl objetivo de este trabajo fue la elaboración de unas recomendaciones para la atención integral de la cistinosis y la transición del adolescente a la medicina del adulto, basadas en la experiencia clínica, con el fin de reducir el impacto de la enfermedad y mejorar la calidad de vida y el pronóstico del paciente.
MétodoBúsqueda bibliográfica y reuniones de consenso de un equipo multidisciplinar de expertos en la práctica clínica con pacientes afectos de cistinosis (Grupo T-CiS.bcn), procedentes de 5 hospitales localizados en Barcelona.
ResultadosEl documento recoge recomendaciones específicas y necesarias para el diagnóstico, tratamiento y seguimiento multidisciplinar de la cistinosis en las siguientes áreas: nefrología, diálisis, trasplante renal, oftalmología, endocrinología, neurología, laboratorio, consejo genético, enfermería y farmacia.
ConclusionesDisponer de un documento de referencia para la atención integral de la cistinosis constituye una herramienta de soporte para los profesionales de la salud que asisten a estos pacientes. Los principales pilares en los que se sustenta son: a) el enfoque multidisciplinar, b) la adecuada monitorización de la enfermedad y control de los niveles de cistina intraleucocitarios, c) la importancia de la adherencia al tratamiento con cisteamina y d) la promoción del autocuidado del paciente mediante programas de educación en la enfermedad. Todo ello conducirá, en una segunda fase, a la elaboración de un modelo de transición coordinado entre los servicios de pediatría y de adultos que contemple las necesidades específicas de la cistinosis.
Cystinosis is a rare systemic lysosomal storage disease which, if not treated, leads to end-stage kidney failure in the first decade of life.1 Its natural history has been transformed thanks to the development of kidney transplantation (KTx) in children2 and the availability of specific treatment with cysteamine, a drug therapy that should be maintained throughout the patient's lifetime.3 As a consequence, patient survival has increased from the first decade of life to beyond the fourth decade and cystinosis has passed from paediatrics to adult medicine.4
The control of cystinosis is complex owing to its severity and multisystemic nature, and the requirement of treatment with several drugs with a very strict dosage schedule. Early diagnosis, prompt cysteamine administration and treatment adherence influence morbidity and prognosis.5,6 Nevertheless, adherence to therapy, which is usually good in children, tends to wane in teenagers and adults.7 Furthermore, when patients reach adult age, they are usually transferred from the paediatric expert centre to a local hospital with limited experience in cystinosis, while the systemic manifestations continue to progress and the disease becomes more complex.8 This phenomenon is seen in other chronic renal diseases that debut in paediatric patients9 and highlights the need to implement transition strategies and promote patient self-care.10
The current cystinosis count in Spain comprises 56 patients treated and followed up at 22 hospitals. Approximately 50% are adults and 16% adolescents; 57% are kidney transplant recipients.7
The working group for the care and transition of cystinosis in Barcelona (T-CiS.bcn) has assembled a group of experts in the disease to establish, as a first step, recommendations for the comprehensive care of cystinosis and the transition of adolescents to adult-care units in our country. This consensus document presents a support tool for health care professionals both involved in and interested in cystinosis. It is focused on reducing disease impact, improving quality of life and prolonging survival, in accordance with the guidelines of the International Society of Nephrology (ISN) and the International Paediatric Nephrology Association (IPNA).10 At a later date, the T-CiS.bcn group plans to create a coordinated model of transition from paediatric to adult care services which will cover the specific needs of cystinosis.
EtiopathogenesisCystinosis is a hereditary autosomal recessive disease caused by mutations with loss of function of the CTNS gene (chromosome 17p13), which encodes for cystinosin.11 Cystinosin is a specific transmembrane protein for the transport of cystine from the lysosome to cell cytoplasm.12 Its absence causes progressive deposits of intralysosomal cystine, the main diagnostic marker of the disease1. Its annual incidence is estimated at 1/100,000–200,000 newborns, while the population prevalence is 1–9/1,000,000.13 The most frequent mutation in the CTNS gene is a deletion of 57Kb14 which is also observed in 34% of patients in the Spanish population.15
The amino acid cysteine oxidises inside the lysosome and forms cystine. In patients with cystinosis, there is an accumulation of cystine that precipitates in crystal form in all the cells of the organism, particularly in renal and ocular tissue.16 The increase in the lysosomal concentration of cystine is associated with increased cellular apoptosis, oxidative stress and alterations in the metabolism of glutathione and arachidonic acid.17–19 Other pathogenic mechanisms involved are inflammatory20 and “endoplasmic reticulum stress”, which finally lead to cell death.21,22
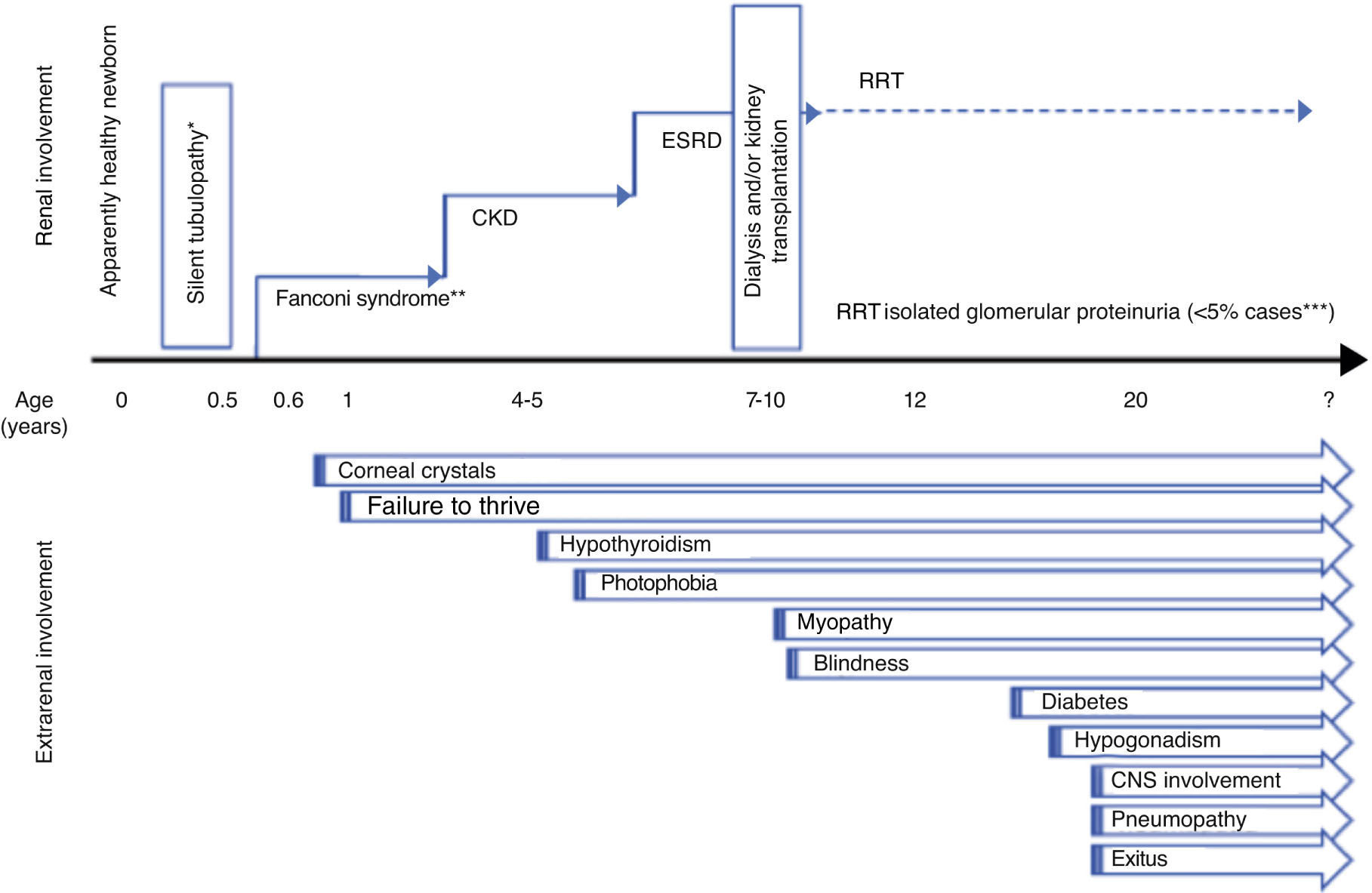
SymptomsCystinosis is a multisystemic disease23 with the kidneys and eyes being the first organs to be affected. Three clinical forms have been described: infantile nephropathic cystinosis (OMIM#219800), the most serious subtype, which debuts early on; juvenile nephropathic cystinosis (OMIM#219900), a less severe subtype, which debuts in childhood or later; and adult non-nephropathic cystinosis (OMIM#219750), with exclusively ocular involvement.24 Nonetheless, in clinical practice, two main subtypes are defined: nephropathic cystinosis that debuts in early childhood with severe Fanconi syndrome (representing 95% of all cases) and late-onset non-nephropathic cystinosis, which appears in young patients or adults with renal and/or ocular involvement (representing <5% of affected patients). In some patients, ocular involvement can precede renal manifestations by several years.25
Renal diseaseFanconi syndromeTypical clinical symptoms include the appearance of severe Fanconi syndrome with evolution to chronic kidney disease (CKD) (Fig. 1). Tubulopathy characteristically becomes evident in the second semester of life after a symptom-free interval.24 Affected newborns are apparently normal, although it is possible to detect urinary alterations (alkalineurine with glycosuria and/or proteinuria) very early preceding symptoms.26 Cystinosis is the most frequent cause of inherited Fanconi syndrome24 and should be considered in the initial differential diagnosis in newborns. Nonetheless, cases have been described of cystinotic patients who debuted with atypical symptoms not suggestive of Fanconi syndrome but of distal tubulopathy, such as nephrogenic diabetes insipidus or Batter syndrome. Thus, the diagnosis of cystinosis should be considered in all newborns with complex tubulopathy, particulary if growth is affected and the patient is anorexic.1 The differential diagnosis should contemplate the possibility of secondary proximal tubulopathy.27,28 The severity of Fanconi syndrome associated with cystinosis requires rigorous treatment that is frequently very complex (Table 1).
Symptomatic treatment of renal disease in cystinosis.1,24,29,31
| Therapeutic aims | Treatment |
|---|---|
| Preserve water balance by replacing losses | Replace water loss according to need (between 1.5 and 6L/day), either orally or by nasogastric tube or gastrostomy, if needed. |
| Reduce polyuria: oral indomethacin (1–3mg/kg/day) | |
| Preserve electrolyte balance by replacing losses | Potassium (between 2 and 10mEq/kg/day) |
| ClNa (1–2mEq/kg/day, with progressive increase in dosage) | |
| Phosphorus (between 1 and 4g/day) | |
| Neutralise acidosis (maintain normal blood pH and serum bicarbonate between 22 and 24mEq/L) | Bicarbonate or citrate at an initial dose of 1–2mEq/kg/day, with progressive increase in dosage |
| Nutritional support | Nutritional assessment with appropriate caloric supplementation according to age and kidney function |
| Treatment of bone mineral disease | Cholecalciferol |
| Calcium supplements | |
| Active vitamin D (1 Alpha-vitamin D, Calcitriol, Paricalcitol, others) | |
| Growth hormone (if indicated) | |
| Others | Carnitine29 (100mg/kg/día) |
| ACEi/ARB antiproteinuric agents (assess tolerance) | |
ACEi/ARB: inhibitors of the renin-angiotensin system.
After the age of two, if no specific treatment is administered, glomerular involvement progress with a drop in the glomerular filtration rate (GFR) and an increase in plasma creatinine starting at the age of 4 to 6, which evolves to advanced CKD.1 Concurrently, Fanconi syndrome usually remits and, consequently, it is possible to reduce water/electrolyte supplements (Table 1). In the absence of specific pharmacological treatment (cysteamine), mean age at the onset of end-stage renal disease (ESRD) is 9.2 years. In more contemporary series including patients who received early treatment with cysteamine, there is a significant delay in evolution to ESRD at the age of 13.5 which has been attributed to better patient control by physicians. Furthermore, in cases with very early diagnosis and treatment, there is a growing percentage of patients who remain in predialysis after adolescence.6
There are also forms of attenuated or late-onset cystinosis that debut in adolescence or in young adults, such as glomerular disease and proteinuria without Fanconi syndrome, although occasionally signs suggestive of proximal tubulopathy are observed. Usually, patients also present ocular manifestations of the disease that can be nearly asymptomatic25 (Fig. 1).
Although renal biopsy is not necessary for diagnosis, it demonstrates non-specific lesions of glomerulosclerosis and other more characteristic signs such as irregularities on the brush border of proximal tubule cells, swan neck deformity and, occasionally, deposits of cystine crystals and giant multinucleated podocytes.2,16,24
DialysisThe renal replacement therapy (RRT) of choice in cystinosis is the kidney transplantation (KTx) since the disease does not recur in kidney grafts. In many cases, however, the limitation of organs or delayed diagnosis results in the start of dialysis. According to the NAPRTCS register, 1.4% of patients <18 years of age who initiated chronic dialysis had cystinosis.30 On the other hand, in the European ESPN/ERA-EDTA Registry, 0.9% of patients <20 years of age and 0.1% of patients >20 years with RRT had cystinosis. In Europe, peritoneal dialysis (PD) was the most frequent initial treatment modality (39.6%), followed by preventive KTx (35.1%). Some 17.9% of patients received haemodialysis (HD).5
Fanconi syndrome can persist after the start of dialysis, which influences the dietetic prescription for water, patient diet and the possible need to administer other medications such as phosphorus chelates. Although the urinary loss of saline and polyuria usually decrease in advanced CKD, the patient may continue to need water/electrolyte supplements and carnitine (Table 1). On rare occasions, the severity of Fanconi syndrome justifies nephrectomy of the native kidneys.31 Moreover, many cystinotic patients on dialysis characteristically present extrarenal involvement requiring the integrated intervention of other specialists (see section on extrarenal involvement), which can be a challenge for nephrologists when treating their patients.32
Kidney transplantationAs previously stated, the RRT of choice in cystinosis is KTx. Since graft cells do not carry the lysosomal defect, the disease does not recur in the transplanted organ. However, it is possible to observe interstitial deposits of cystine crystals, which represent leukocytes of the recipient and have no pathological significance.21 Family-donor transplantation is also curative, and heterozygous carriers of the CTNS mutation can be appropriate donors since they do not have the disease.4,6,33
Indirect data from cystinotic patients with advanced kidney failure and international registries suggest that preventive kidney transplantation would be beneficial in this disease, particularly when a living donor is available4 thereby avoiding the need for dialysis.5 Thus, the indication for kidney transplantation is established when the GFR is <20ml/min/1.73m2, which would be somewhat earlier than in other kidney diseases.1
In the United States (USRDS2013), mean age of patients with cystinosis at the first KTx is 13.8 years (range: 2 to 24), which has not changed in recent decades; 32.4% underwent preventive KTx.34 Similarly data from the European Registry (ESPN/ERA-EDTA Registry) show a similar percentage of 35.1% predialysis transplantations, a much higher percentage than in other nephropathies (17.1%).5 Eighty-five percent of all the cystinotic patients in RRT were kidney transplant recipients. Regarding the type of donor, 54% of patients in the US received a living and 46% a deceased donor organ.34 Similarly, in Europe 48.9% received a living donor transplant.5
It is worthy of mention that the duration of functioning kidney grafts in cystinotic patients is longer than that observed in the population of recipients transplanted for other causes.5,35
Extrarenal diseaseThe longer survival and better prognosis of patients with cystinosis have resulted in better understanding of the multiple organ involvement in this disease4,6,32,36 (Fig. 1).
Ocular involvementEye involvement is intrinsic to cystinosis. The presence of cystine crystals in the cornea is a diagnostic criterion for this disease,37 although its absence before the age of 12 months may not rule it out.1
Cystine deposits in the cornea are one of the earliest manifestations of cystinosis (Fig. 1). Although no crystals are present at birth, they can be observed in children who are only a few months old.1 Initially, they are deposited in the superficial layers of the peripheral cornea, but progressively begin to affect all the layers and entire extension of the cornea. If left untreated, corneal crystal deposits progress inexorably, increasing with age, resulting in photophobia, which can be quite incapacitating, and in abnormal corneal sensitivity. In time, this condition leads to recurrent corneal erosions and stromal oedema, which can reduce visual acuity. Reports also exist of calcium deposition in Bowman's membrane or band keratopathy, which affect vision when situated in the visual axis.37
Crystals are also deposited in other ocular structures such us the conjunctiva, anterior chamber, iris, ciliary body, choroid and retina. Retinal involvement causes degeneration of the photoreceptors, mainly the rods, thereby altering the peripheral visual field and night vision, although central vision may also be reduced. Less frequently, there have been reports of posterior synechiae, adherence of the iris to the anterior lens capsule and neovascularisation of the peripheral cornea.38,39 Furthermore, tear production is reduced, causing dry eye, and neuro-ophthalmological manifestations (papilledema and ophthalmoplegia) are observed secondary to the increased intracranial pressure reported in this disease.4 In late-onset disease, the presence of crystals might not be detected until adulthood.25
Growth and development: mineral-bone involvementFailure to thrive is a classic clinical symptom of cystinosis and frequently is the reason for an early consultation.36 The underlying mechanism is multifactorial, although it is related to Fanconi syndrome severity. The concurrence of metabolic acidosis, hyponutrition, increased gastrointestinal and renal losses and CKD lead to delayed growth that can be very severe.40,41 Similarly, patients present endocrine alterations (see endocrine involvement section) and, infrequently, primary growth hormone (GH) deficiency.42
Patients with inadequate treatments usually have shorter stature.36 Classically, the adult height reported in patients with suboptimal treatment is 144cm and weight 45kg (25cm and 25kg below the normal population average, respectively).4 The most recent series with better therapeutic control reported less retarded growth8 and a favourable impact of treatment on growth regulation mechanisms.43 Nevertheless, nowadays 27% of transplanted cystinotic patients and 44% of those on dialysis continue to be shorter than average.5
Early GH administration improves height and weight, although the therapeutic response is usually lower than that observed in CKD due to other causes, despite optimum disease control. GH is an essential therapeutic tool in this disease, for its impact on longitudinal growth and its anabolic effect.40,44 Patients with cystinosis develop a characteristic metabolic bone disease caused by different factors: bone deposits of cystine crystals, mineral deficiency, renal rickets24 and CKD per se.45 Bone anomalies attributed to copper deficiency, possibly secondary to Fanconi syndrome have also been described.46 It is therefore common to detect osteopenia, especially in transplant recipients, related also to other endocrine alterations of the disease (see endocrine involvement section) and potentially to the treatment.23,47 Some patients have bone fragility and a higher risk of fractures.32
Endocrine involvementEndocrine manifestations are caused by destruction of the affected glands due to cystine deposits; their incidence and age at appearance are associated with the establishment of specific treatment with cysteamine.43
Primary hypothyroidism is the most frequent endocrine complication;23 it is progressive and requires chronic treatment with levothyroxine.1,4 Diabetes mellitus (DM) is characterised by a progressive alteration in insulin secretion,48 with negative immunology, and requires treatment with insulin.2 It is observed in transplant recipients who receive corticosteroids.23 In males, cystinosis causes primary hypogonadism and infertility is a constant.4,49 In females, however, neither hypogonadism nor infertility are prevalent, and affected patients can thus have children,50 although the risk of prematurity is increased.51
Cardiovascular involvementThe appearance of dyslipidemia and vascular calcification due to cystinosis and CKD are considered increased cardiovascular risk factors.23,32,41 42% of patients develop arterial hypertension, usually post-transplantation. Aortic aneurysms and coronary vessels involvement, as well as cardiomyopathy associated with cystine crystals deposits in the myocardium have also been reported.36 In adult patients, screening for ischaemic heart disease is recommended.4
Neurological involvementCystinosis is associated with alterations in cerebral structure and increased cystine levels in different areas of the nervous system and muscle tissue.4,6,32,52 In general, neurological complications worsen the prognosis of the disease:
- •
Progressive ischaemic myopathy4,32,53 is predominantly distal and begins in the hands; loss of muscle mass is also observed with later ventilator capacity impairment and swallowing difficulties. Some authors attribute muscle weakness in these patients to carnitine deficiency.24,28,29
- •
Central nervous system (CNS) involvement4,31 is mainly observed in patients with suboptimal treatment with cysteamine:
- •
Neurocognitive alterations:67–73 in cystinotic patients, a specific profile of alterations in visual-motor integration, visual memory, maintained attention, planning, motor processing speed and arithmetic calculation have been described. Consequently, they account for a significant incidence of social difficulties that could explain the behavioural phenotype in some patients. Intelligence is usually normal.
Early detection of neurological complications in cystinosis facilitates better therapeutic strategies, reduces the number of hospitalisations and improves quality of life. The participation of a neurologist will help to evaluate the functional capacity of patients and detect earlier the neurological manifestations that can affect autonomy in basic daily life activities.8,31,32,55
MiscellaneousThe ubiquity of cystinosis is demonstrated by its non-specific symptoms e.g. gastrointestinal and other disorders such as heat intolerance and hypophoresis. Similarly, the systemic nature of the disease explains the progressive appearance of other clinical symptoms secondary to the deposition of cystine crystals in different organs and systems, as detailed below (Fig. 1):
- •
Digestive system74:
- –
Nausea, vomiting, epigastralgia, anorexia
- –
Increased gastrin secretion (associated with taking cysteamine)
- –
Decreased salivation
- –
Mechanical swallowing difficulties
- –
Delayed gastric emptying and intestinal and intestinal dysmotility
- –
Intestinal pseudo-obstruction
- –
Intestinal inflammatory disease
- –
- •
Liver32,75:
- –
Regenerative nodular hyperplasia without liver failure
- –
Non-cirrhotic portal hypertension with hypersplenism
- –
Cholestasis
- –
Hypercholesterolemia
- –
- •
Skin1,76:
- –
Hypopigmentation of skin and hair due to altered melanogenesis
- –
Altered sweating and intolerance to heat
- –
- •
Bone marrow4:
- –
Anaemia
- –
Coagulopathy due to dysfunctional platelets
- –
The clinical diagnosis of cystinosis is symptom-based and is confirmed by biochemical and molecular diagnosis.
Clinical diagnosisGuiding signs are early-onset of severe Fanconi syndrome and the detection of corneal crystals. As the disease progresses, systemic involvement may be observed (Fig. 1). However, in patients with less severe forms, renal involvement is restricted to proteinuria and CKD. Occasionally, corneal crystals in adult patients with CKD of unknown aetiology lead to the diagnosis of cystinosis.25 Kidney biopsy, while not a requirement for the diagnosis of cystinosis, can be useful in these atypical presentations.2,16,24,32
General biochemical diagnosisThis diagnosis is based on the detection of water/electrolyte disorders, affected acid-base balance and eventually renal function, which are all prototypical of Fanconi syndrome.24,28,29
Specific biochemical diagnosisThis involves the detection of elevated white blood cells (WBC) cystine levels.77 Currently High Performance Liquid Chromatography–Tandem Mass Spectrometry (HPLC-MS/MS) in granulocytes is used as it is a more sensitive technique.78,79
The reference values are:
- •
healthy individual: ≤0.5nmol 1/2 cystine/mg protein (values >0.5 may have diagnostic significance, and it is recommended to repeat the determination)
- •
affected individual without treatment: >1nmol 1/2 cystine/mg protein (usually >2)
- •
individual treated with good therapeutic control: ≤1 nmol 1/2 cystine/mg protein
Normal WBC cystine levels in newborns do not completely rule out the diagnosis. Therefore, in cases where cystinosis is highly suspected, a second test is recommended 3–6 months after the first study when the results are not conclusive79 (Table 2).
Biochemical diagnosis and WBC cystine level monitoring. Molecular diagnosis (www.orpha.net).
| Requirements for WBC cystine level monitoring77–79 | |
|---|---|
| Sampling conditions | No fasting required |
| In treated patients, blood extraction should be made 6h post-cysteamine dose | |
| Collect within lithium or sodium heparin tube | |
| Ship immediately to the laboratory: it should be processed into 24 hours post-sampling | |
| Keep at room temperature | |
| Minimum volume | For patients <10kg body weight: 6mL blood |
| For patients ≥10kg body weight: 10mL blood | |
| RECOMMENDATIONS for monitoring WBC cystine levels | At the start of Cystagon® treatment |
| Monthly after dose adjustments | |
| Every 6 months in stable patients | |
| Increase frequency in cases of significant clinical changes (Transplantation and Dialysis) | |
| Reference Laboratory in Spain | |
| Hospital Clínic de Barcelona | |
| Servicio de Bioquímica y Genética Molecular. Sección de Errores Congénitos del Metabolismo | |
| Dra. Judit García Villoria: jugarcia@clinic.ub.es | |
| Tfn. 93 227 56 00 Ext. 7585 | |
| C/ Mejía Lequerica s/n. Edificio Helios III. Planta baja. 08028 Barcelona, Spain. | |
| Requirements for the molecular diagnosis ofCTNSgene12,13,15,80 | |
| Genetic testing of patient and family | |
| Sample | 2–3mL blood in EDTA at room temperature |
| DNA at room temperature | |
| Prenatal testing | |
| Sample | DNA from cultured amniocytes or chorionic villi sampling |
| Prior identification of the disease-causing mutations in parents and in index case | |
| Previous appointment with the laboratory is essential | |
| Reference Laboratory in Spain | |
| Hospital Clínic de Barcelona | |
| Servicio de Bioquímica y Genética Molecular. Sección de Errores Congénitos del Metabolismo | |
| Dra. MªJosep Coll: mjcoll@clinic.ub.es | |
| Tfn. 93 227 9341 | |
| C/ Mejía Lequerica s/n. Edificio Helios III. Planta baja. 08028 Barcelona, Spain. | |
Cystinosis is confirmed by the detection of homozygous or compound heterozygous mutations in the CTNS gene. More than 100 different mutations have been reported and the most frequent one is the ∼57kb deletion affecting the first 10 exons, especially in patients of Northern European descent. Specific mutations turn into an absence of protein or a probably non-functional truncated protein11,12,14,15,80 (Table 2).
Genetic counsellingSince cystinosis is an autosomal recessive disease, the probability of a family with one affected child having another with cystinosis is 25%.81 In this case, genetic counselling includes information on prenatal diagnostic techniques and embryo selection.82,83 The probability of a woman with cystinosis having an affected child is very low, except in consanguineous families or endogamous populations. Males with cystinosis are universally sterile.49
Genetic counselling usually includes information on patient associations84–86 and institutional strategies in rare diseases.87
TreatmentSymptomatic treatment of kidney involvementThe aim of treatment (Table 1 & Annex 1) is to control Fanconi syndrome, its complications and other factors involved in kidney failure progression.24–28 In ESRD, promoting KTx is a priority. Regardless of kidney involvement, all patients should receive specific treatment with cysteamine for the prevention and therapeutic control of the systemic disease.
Treatment of CKD should follow international guidelines.88–90 In transplanted patients, minimising or avoiding corticosteroids use is recommended.23
Specific treatment with cysteamineOral cysteamineThe specific treatment for all clinical forms of cystinosis is oral cysteamine. Cysteamine depletes lysosomal cystine content by forming disulphide cysteine–cysteamine complexes able to exit the lysosomes by means of the alternative lysine channel, and the remaining cysteine via a cysteine carrier.3,19,91
The first specific pharmacological treatment for cystinosis is Cystagon® (oral cysteamine bitartrate in hard capsules), the only authorised therapy in Spain92–94. A new formula in hard gastro-resistant capsules has recently been approved for cysteamine.93–96
Therapeutic benefitsOral cysteamine should be initiated at the time of diagnosis and continued lifelong. When compliance is consistent, cysteamine is able to deplete up to 95% of cellular cystine deposits.62 The reduction in these deposits correlates with cystinosis severity.32 It has been demonstrated that cysteamine prolongs the life of the patient, while delaying kidney disease progression and the need for renal replacement therapy.5,97 Similarly, it reduces the severity and frequency of extrarenal complications.32 Prognosis of the disease is directly related to early treatment and its duration. Even when cystinosis diagnosis is delayed, cysteamine has demonstrated clinical benefits.4,6,98 Although Fanconi syndrome is not usually reversible with cysteamine,19 in some isolated cases of prenatal diagnosis the beginning of cysteamine therapy within the first weeks of life prevented the appearance of tubulopathy.99,100
Cystagon®: dosage, administration and treatment monitoringTreatment is based on the depletion of lysosomal cystine content which, in clinical practice, means the reduction of WBC cystine levels, with an optimal therapeutic goal <1nmol hemicystine/mg of protein. The decrease in those WBC cysteine levels correlates with plasma cysteamine concentrations for the 6 hours following Cystagon® dose, being minimum at ∼2h after the drug is taken and returning to baseline (pre-dose) 6h later. This explains the need to take the drug every 6h, overnight dose included101,102 (Table 3).
Specific treatment with cysteamine.
| Oral Cysteamine – Cystagon®31,47,92–94,102 | ||
|---|---|---|
| Dosage | ||
| By age | Children ≤12 years | Patients >12 years |
| Recommended dosage | by body surface area (g/m2/day) | if weight > 50 Kg |
| (Dosage should be divided 4 times daily) | 1.30g/m2/day | 2g/day |
| Starting dosage | 1/4 to 1/6 of the recommended dosage | |
| Increase gradually over 4–6 weeks to avoid intolerance** | ||
| Dosage adjustments | Raise if adequate tolerance and WB C cystine level is greater than > 1nmol ½ cystine/mg protein | |
| Maximum dosage | 1.95g/m2/day | |
| Overdosing is not recommended since it does not improve the prognosis and is associated with adverse effects47 | ||
| Renal insufficiency | No dose adjustment required | |
| RECOMMENDATIONS for appropriate dosage | Treatment should be initiated under the supervision of a physician experienced in the treatment of cystinosis | |
| 4 doses per day; every 6 hours; night dose included | ||
| Adjust the dose according to WBC cystine levels | ||
| Hard capsules should not be administered to children under the age of 6 years owing to risk of aspiration | ||
| If needed, open the capsules and sprinkle the content on food | ||
| Cysteamine powder could be mixed with milk, potatoes and other starch-based products. Avoid acidic drinks. | ||
| CYSTAGON® should be restarted after renal transplant to prevent non-renal complications | ||
| Side effects | Gastrointestinal disorders due to gastric acid hypersecretion | |
| RECOMMENDATIONS for improving gastrointestinal tolerability: | ||
| Concomitant administration of proton pump inhibitors102 | ||
| Administer with meals or immediately afterwards. Intake is recommended with food such as milk, potatoes or other starchy foods92–94 | ||
| Characteristic body odour and halitosis | ||
| RECOMMENDATIONS for improving halitosis: mentholated pills31 | ||
| Other adverse reactions refer to SMPC92–94 | ||
| Drug interactions | No interaction studies have been conducted | |
| Can be administered in conjunction with electrolytic or mineral supplements, vitamin D analogues, tyrosine or immunosuppressive drugs | ||
| Cysteamine eye drops37–39,105–109 | ||
| Eye drops | 0.55% cysteamine solution (see Annex 1) | |
| Dose | 1 drop/eye | |
| Dosage | 10–12 instillations/day | |
Monitoring of WBC cysteine levels at the start of treatment and monthly after changes in the prescribed dosage is recommended. In patients with maintained cystine levels, follow-up controls are recommended every 6 months. Similarly, in an individualised manner, the frequency of monitoring should be increased in case of significant clinical changes such as KTx and dialysis77,78,79 (Table 2).
Oral treatment in special situationsChronic kidney disease, dialysis and transplantationSince there is no correlation between GFR and plasma cysteamine levels, it is not necessary to adjust the dosage to renal function; instead, the prescribed dosage should be adjusted to the quantification of WBC cystine levels. Adjustments for Fanconi syndrome are also unnecessary.6,103
Pregnancy and breastfeedingAlthough data is insufficient, reproductive toxicity and teratogenic effects of cysteamine have been observed in animals.104 Its use is therefore contraindicated during pregnancy, particularly in the first trimester. Family planning is recommended in women of childbearing age. Furthermore, cysteamine administration should be avoided during breastfeeding.
Cysteamine eye dropsSpecific treatment of ocular involvement in cystinosis requires, in addition to oral cysteamine, the use of cysteamine eye drops. The ophthalmological therapeutic strategy32,37 makes a distinction between:
Corneal involvementCystine crystal deposits should be treated with topical cysteamine since the cornea is an avascular structure and, consequently, oral medication is not effective for the cornea.37,38,105,106 The recommended prescription is shown in Table 3. Viscous formulae are being developed to achieve longer contact of cysteamine with the ocular surface and be able to reduce the frequency of instillations with equal efficacy.107,108
Involvement of non-corneal structuresOral cysteamine is effective in the retina and other ocular structures. The incidence of retinopathies has decreased with the systemic use of cysteamine. The frequency and severity of non-corneal manifestations are directly related to compliance with oral cysteamine treatment, and the risk of important vision loss may arise if systemic treatment is not correctly followed.39,109
Compliance with specific cysteamine treatmentThe World Health Organization (WHO) defines compliance as the degree to which patient behaviour follows the recommendations of medical professionals.110
The impact of non-adherence in cystinosis results in poorer prognosis and faster progression of the renal and extrarenal disease in patients who are non-compliant compared with those who are.5,6,97
Information on adherence in patients with cystinosis is limited, although monitoring WBC cystine levels is able to detect non-compliant patients.77,79 Other studies have confirmed adequate adherence to Cystagon® in children patients which wanes significantly in adolescents and adults.7,8 Nonetheless, in groups of highly motivated patients, only 8% had compliance problems.111
In cystinosis, risk factors for non-compliance with cysteamine therapy include: dosage schedules, problems with tolerance, side effects and the requirements of several medications for the control of the clinical manifestations of the disease. Moreover, other risk factors which are not exclusive to cystinosis are: limited knowledge of the disease, lack of motivation, inadequate transition of patients to adult care units and impact of the disease on quality of life.7,9,10
Nevertheless, suboptimal treatment compliance is not a phenomenon restricted to cystinosis. Recent studies report that 52 to 67% of adult kidney transplant recipients do not correctly follow prescribed immunosuppressant treatment, which increases the probability of graft loss.112,113 These percentages are similar to those published in patients with cystinosis in our population,7 which may indicate the coexistence of scenarios in common with CKD. Therefore, in order to improve adherence in cystinotic patients, the recommended strategy is to correct risk factors for non-compliance and promote patient self-care, similar to the strategies used successfully in adult kidney transplant recipients114–119 (Table 4).
Recommendations for improving treatment compliance.7,9,10,114–119
| Identify risk factors that affect adherence and apply corrective measures when possible: |
| Intrinsic patient and socio-economic factors |
| Disease-related factors |
| Treatment-related factors |
| Healthcare system organisation barriers |
| Identify and assign a “Patient health Coordinator” |
| Promote patient education and treatment support: |
| Implement disease education programmes |
| Establish treatment programmes: easy to follow with support measures for compliance |
| Use questionnaires to detect non-compliance |
| Follow-up of appointments and absences |
| Develop patient support programmes involving family members, friends and patient associations |
| Create a multidisciplinary medical team |
| Implement protocols for transition to adult care |
T-CiS.bcn group members, after an exhaustive review of medical literature and based on our clinical practice with patients, have stablished recommendations for the multidisciplinary care and connected transition from paediatric to adult-care units in cystinosis. Our aim was to provide support tools and medical advice to health care professionals involved and interested in the care of cystinosis.
These recommendations are presented in Tables 4–8.
Recommendations for follow-up of patients on renal replacement therapy (RRT): dialysis (D) and transplantation (TxR).
| Recommendations for dialysis6,103 |
|---|
| Promote preventive KTx as an initial method of RRT in patients with advanced CKD |
| Monitor residual urine volume and urinary saline loss to adapt the dialysis prescription and avoid excessive ultrafiltration |
| Maintain the general treatment of Fanconi syndrome and adapt the diet in an individual manner |
| Carefully monitor extrarenal involvement on a multidisciplinary basis |
| Oral and ophthalmological cysteamine treatment must be maintained |
| Cysteamine dosage should not be adjusted to glomerular filtration rate (see Specific Treatment with Cysteamine section) |
| Recommendations for kidney transplantation4–6,33,88–90 |
| Prior to waiting list inclusion |
| Promote preventive kidney transplantation when the glomerular filtrate ≤20mL/min/1.73 m2 |
| Living or deceased donor |
| Evaluate the associated Fanconi syndrome (residual diuresis – can be very high – saline loss, rickets, tubular acidosis, carnitine deficiency) |
| Monitor WBC cystine levels and optimise treatment with cysteamine (oral and ophthalmological) |
| Assess possible systemic involvement and its impact on the transplant (hypothyroidism, diabetes, cardiovascular disease, bone disease, swallowing disorders) |
| Prescribe fluids and individualised diet. Assess phosphate, potassium, bicarbonate and carnitine supplement requirements |
| Pre-transplant and peri-transplant |
| Avoid volume depletion before and during surgery (intensive endovenous fluid therapy to guarantee normovolaemia, including potassium and bicarbonate supplements) |
| Immunosuppression according to hospital protocol |
| Temporary suspension of cysteamine treatment |
| Immediately post-transplant |
| Administer fluid therapy and sufficient electrolytes to maintain adequate water/electrolyte balance and good control of residual Fanconi syndrome |
| Monitor the possible appearance of diabetes. |
| Immunosuppression therapy according to hospital protocol |
| Continued post-transplant care |
| Reintroduce cysteamine once the patient and graft are stable, approximately 3–4 weeks post-transplantation, at increasing doses up to therapeutic doses |
| Monitor WBC cystine levels |
| Immunosuppression following hospital protocol; promote reduction in and/or suspension of corticosteroids |
| Controls and follow-up in accordance with recommendations and clinical guidelines |
| Maintain ophthalmological treatment with cysteamine and stimulate treatment compliance |
| Assess possible systemic involvement and its impact on transplantation. Promote and standardise a multidisciplinary health care plan for cystinosis |
In any clinical situation, it is necessary to administer specific cystinosis treatment with oral cysteamine to maintain recommended WBC cystine levels <1nmol hemicystine/mg protein and cysteamine eye drops to eliminate corneal deposits (see Table 3).
Recommendations for ophthalmological follow-up.4,37–39,105–109,120
| Test | Ocular structure | Frequency | Observations |
|---|---|---|---|
| Slit lamp biomicroscopy | Study of the cornea and rest of anterior segment | Annual | |
| Intraocular pressure measurement | Rule out ocular hypertension | Annual | |
| Dilated fundus examination | Assess optic disc and retinal pigmentation | Annual | Urgent: if the patient reports severe loss of VA (uncommon) |
| In patients with GH, do a baseline assessment and after 4 months to detect intracranial hypertension120 | |||
| Photopic and scotopic ERG | Functionality of rods and cones | Only if patient reports altered night vision or abnormal retinal examination | |
| Equipment | Slit lamp* | ||
| Tonometer | |||
| Indirect ophthalmoscope | |||
| In any clinical situation, specific cystinosis treatment is required with oral cysteamine to maintain recommended WBC cystine levels <1nmol hemicystine/mg protein and cysteamine eye drops to eliminate corneal deposits (See Table 3). | |||
ERG: electroretinogram; GH: growth hormone; VA: visual acuity.
Very sensitive for diagnosing cystine crystals in the cornea, but less useful for patient follow-up since the quantification of crystals is rather subjective. It is therefore interesting to record detailed data at each examination on the distribution of crystals in the cornea, specifying whether the deposition is only peripheral or diffuse and whether they are located in the epithelium, stroma and/or endothelium.
Recommendations for the follow-up and treatment of endocrine system involvement.4,5,32,48,49,121
| Disorder | Complementary studies | Frequency | Treatment | Observations |
|---|---|---|---|---|
| Hypothyroidism | TSH, T4L, Anti-thyroid Ab/Imaging study not necessary | At diagnosis | Levothyroxine | Initiate treatment when TSH > 10 mIU/L/if there are symptoms, consider initiating with TSH, 5–10 mIU/L |
| TSH, T4L | Routine follow-up: Quarterly for dosage adjustments/annual, if TSH is normal | |||
| Diabetes Mellitus | Glycaemia, HbA1c (optional: c-peptide) | Diagnosis | Insulin | If pancreatic reserve is sufficient, consider single daily dose of slow-release insulin |
| If symptoms of polyuria-polydypsia: laboratory tests with ionogram, venous blood gases and ketonuria | ||||
| Glycaemia, HbA1c | Quarterly or biannual follow-up, according to clinical criteria/annual, if no symptoms | |||
| Lipid profile: total cholesterol, LDL, HDL and TG | Annual | Dyslipidaemia treatment | ||
| Funduscopy | Annual | According to ophthalmologist criteria | ||
| Pain and vibration sensitivity | Annual | Education to prevent lesions | ||
| Examination feet and pulse | Annual | |||
| Impaired longitudinal growth | Nutritional assessment | According to clinical criteria | Optimise nutrition | Consider supplements and referral to nutritionist |
| Height and weight | At each visit | Until bone maturity | ||
| See Fanconi syndrom section | Treatment of Fanconi syndrome | |||
| Bone age | Diagnosis | r-GH | In pubescent patients, rule out hypogonadism/in transplant recipients, consider withdrawal or reduction of corticosteroids | |
| GH, IGF-1, IGFBP-3 | ||||
| If no kidney failure: GH secretion studies | ||||
| Bone age, IGF-1 | Annual follow-up | |||
| Funduscopy | Before initiating GH and after 3–4 months under treatment | Intracranial hypertension secondary to GH usually occurs at the start of treatment (mean: 3–4 months) | ||
| Urgent if headache or reduced vision | ||||
| Calcium, phosphorus, alkaline phosphatase, 25OH·D3, PTH | Annual | Vitamin D, if deficiency | Rule out rickets and vitamin D nutritional deficiency | |
| Calcitriol, if kidney failure | ||||
| Bone mineral density | In adulthood; to be assessed according to results. | If there is osteoporosis, consider specific treatment | ||
| Hypogonadism | Physical exam: sexual maturation and secondary sexual characteristics | At each visit | During puberty period until complete maturity | |
| Testosterone, SHBG, LH, FSH+assessment of thyroid function (see above) | Diagnosis | Daily testosterone replacement therapy (TRT): Topical (if panic to injections), or IM (if compliance problems) | If low testosterone with normal LH and FSH levels: MRI of pituitary | |
| T, LH, FSH | Annual follow-up | |||
| Fertility | Semen analysis | Diagnosis | Infertility is a constant feature in affected males | |
| Consider high-risk pregnancy | Suspend oral cysteamine during gestation | |||
| In any clinical situation, specific cystinosis treatment is required with oral cysteamine treatment to maintain recommended WBC cystine levels <1nmol hemicystine/mg protein and topical cysteamine to eliminate corneal deposits. | ||||
25OHD3: 25-hydroxycholecalciferol (calcifediol); FSH: follicle-stimulating hormone; GH: growth hormone; HbA1c: glycated haemoglobin; IGF-1: insulin-like growth factor 1; IGFBP3: insulin-like growth factor-binding protein 3; LH: luteinising hormone; PTH: parathyroid hormone; rGH: recombinant growth hormone; SHBG: sex hormone-binding globulin; T: testosterone; T4: free thyroxine; TG: triglycerides; TSH: thyroid-stimulating hormone.
Recommendations for the follow-up and treatment of neurological involvement.4,52–73,122–126
| Disorder | Evaluation | Complementary studies | Frequency | Treatment | Observations |
|---|---|---|---|---|---|
| Motor functions of the skeletal muscles4,52,53 | Degree of muscle atrophy/disease progression/degree of disability | MRC scale | Annual | Rehabilitation | Use validate instruments |
| Quantitative determinations of hand muscle strength (Jamar dynamometer, Martin vigorimeter and Jamar Hydraulic Pinch Gauge) | Annual | ||||
| Electromyography | According to clinical criteria | ||||
| Muscle MRI | According to clinical criteria | ||||
| Muscle biopsy | According to clinical criteria | ||||
| Orofacial motor function (language) and swallowing58,122,123 | Facial and bulbar muscles: strength and range of movement of lips, tongue, soft palate, jaw and facial muscles in phonation, articulation, swallowing, breathing and expressions | Focused physical examination | Annual | Reeducation if early signs of dysphagia are detected to prevent bronchoaspirates | |
| Video fluoroscopy | According to clinical criteria | Objective: to asses different phases of swallowing and movement of food bolus within the oral cavity and its passage through the esophagus | |||
| Respiratory muscle function124 | Presence of dyspnoea, apnoea and/or snoring during sleep, morning headaches, daytime hypersomnia, decreased coughing capacity or increased anomalies in expectoration | Spirometry | Annual | Symptomatic respiratory physiotherapy, ventilatory support (CPAP/BIPAP) | Especially in patients with swallowing disorders, risk of bronchoaspirtion or who present symptoms of neuromuscular respiratory insufficiency |
| Oxygen saturation Arterial blood gas | Annual | ||||
| Polysomnography | Annual | ||||
| Central nervous system54–57,59–68,125,126 | Signs of involvement (headache, episodes of unconsciousness, poor school performance, decline in cognitive functions, behavioural alterations, etc.) | Focused physical examinationNeurophysiological studiesNeuroimaging | AnnualAccording to clinical criteriaAccording to clinical criteria | Directed at underlying disorder | Complementary tests if anomalies are detected |
| Neurocognitive alterations69–73 | Neuropsychological examination | Specific evaluation questionnaires for overall cognitive performance, attention and executive functions, language, memory; perceptive, visuospatial and visuoconstructive functions; voluntary cognitive motor control | Annual | Directed to underlying disorder | Adapted to patient's age (school children and adults) |
| In any clinical situation, specific cystinosis treatment is required with oral cysteamine treatment to maintain recommended WBC cystine levels <1nmol hemicystine/mg protein and topical cysteamine to eliminate corneal deposits. | |||||
MRI: magnetic resonance imaging, CPAP: Continuous Positive Airway Pressure, BIPAP: biphasic positive airway pressure.
The authors have no conflicts of interest to declare.
| Drug | Brand name/Product | Content |
|---|---|---|
| Alkaline supplements | ||
| Sodium bicarbonate | Sodium bicarbonate Torres Muñoz 500mg (30 cap.) | 500 mg/comp. |
| Sodium bicarbonate Torres Muñoz 60 g; 200 g; 750 g (powder) | ||
| Sodium bicarbonate Serra 180 g (powder) | ||
| Sodium bicarbonate Viviar 210 g; 250 g; 500 g (powder) | ||
| Sodium bicarbonate NM 1 g; 2 g (sachet) | 1g/sachet; 2g/sachet | |
| *Sodium bicarbonate 1M (8,4%) solución oral | 1mEq/mL | |
| Sodium citrate | *Bicitra oral solution | 1mEq Bicarbonate/mL; 1mEq Na/mL; 0.5mmolcitrate/mL |
| Potassium citrate | *Polycitra oral solution | 2mEq Bicarbonate; 1mEq Na/mL; 1mEq K/mL; 1mmolCitrate/mL |
| *Polycitra LC with phosphrous-oral solution | 2mEq Bicarbonate; 1mEq Na/mL; 1mEq K/mL; 1mmolCitrate/mL; 0.6mmolP/mL | |
| Potassium supplements | ||
| Potassium ascorbate | Boi-K (20 effervescents tablets) | 1mEq K/tablet |
| Bok-K Aspártico (20 effervescents tablets) | 25mEq K/tablet | |
| Potassium chloride | Potasion 600 mg (60 cap.s) | 8mEq K/tablet |
| Potassium glucoheptonate | Potasion 1,32 g/5 mL (125 mL; 250 mL) | 1mEq K/mL |
| Phosphorus supplements | ||
| Sodium Phosphate | *Phosphorus oral solution or Joulie Solution | 1mmol P/ml (30.9mg P/ml) |
| Phosphate Sandoz 500mg (100 cap.) | 16.1mmol P/comp (500mg P/comp.) | |
| Sodium Phosphate monobase NM (100 sachets) | 26mmol P/sobre (800mg P/sachet) | |
| *Polycitra LC with phosphrous-oral solution | 2mEq Bicarbonate; 1mEq Na/mL; 1mEq K/mL; 1mmol Citrate/mL; 0.6mmol P/mL) | |
| Others | ||
| Carnitine | Carnicor 1.5g/5mL (40mL oral solution) | 300mg/mL carnitine |
| Carnicor 1g drinkable vials (10mL) | 100mg/mL carnitine | |
| Secabiol 300mg/mL (40mL oral solution) | 300mg/mL carnitine | |
| Indomethacin | Artinovo 25mg (30 cap.) | 25mg indomethacin |
| Flogoter 25mg (40 cap.) | 25mg indomethacin | |
| Inacid 25mg (30 cap.) | 25mg indomethacin | |
| Indonilo 25mg (24 cap.) | 25mg indomethacin | |
| *Indomethacin 2mg/mL oral solution | 2mg indomethacin | |
| Cysteamine | *Cysteamine 0.55% eye drop solution | 0.55% cysteamine |
cap.: capsules, *: magistral formulation.
| Compounded formulations | ||
|---|---|---|
| Liquid formulation | Component | Amount |
| Sodium bicarbonate 1M (8.4%) oral solutiona | Sodium bicarbonate | 8.4g |
| Sterile distilled water (qs) | 100mL | |
| Bicitra oral solutionb | Sodium, citrate 2H2O (Tri-) | 10g |
| Monohydrated citric acid | 6.7g | |
| Simple syrup with preservatives | 50mL | |
| Sterile distilled water | 40mL | |
| Polycitra oral solutionb | Potassium, citrate H2O (Tri-) | 11g |
| Sodium, citrate 2H2O (Tri-) | 10g | |
| Monohydrate citric acid | 6.7g | |
| Simple syrup with preservatives | 50mL | |
| Sterile distilled water | 38mL | |
| Polycitra LC with phosphorus, oral solutionc | Sodium hydrogenophosphate-12 | 1.4g |
| Phosphoric acid 85% | 1.4mL | |
| Sterile distilled water | 32mL | |
| Potassium, citrate H2O | 11g | |
| Sodium, citrate | 10g | |
| Monohydrate citric acid | 6.7g | |
| Simple syrup (qs) | 100mL | |
| Orange extract | 1 drop | |
| Indomethacin 2 mg/mL oral solutiond,e,f,g | Indomethacin | 0.2g |
| Ethyl alcohol | 0.7mL | |
| Sterile distilled water | 0.3mL | |
| Simple syrup+Nipagin/nipasol | 100mL | |
| Phosphates, oral solution (Joulie solution)b | Phosphoric acid 85% | 5.45g |
| Disodium phosphate 12 H2O (di) | 18.72g | |
| Sterile distilled water (qs) | 100mL | |
| Cysteamine 0.55% eye drop solutionh | Benzalkonium chloride | 0.045g |
| Sodium chloride 0.9% | 225mL | |
| Cysteamine hydrochloride | 1.2375g | |
Trissel LA. Trissel's Stability of Compounded Formulations. 3rd Ed. American Pharmacists Association, Washington DC; 2005:388–389.
The United States Pharmacopeial convention. USP-Pharmacists’ Pharmacopeia, 2nd Ed., Rockville MD; 2008.
Please cite this article as: Ariceta G, Camacho JA, Fernández-Obispo M, Fernández-Polo A, Gamez J, García-Villoria J, et al. Cistinosis en pacientes adolescentes y adultos: recomendaciones para la atención integralde la cistinosis. Nefrologia. 2015;35:304–321.



