


Idiopathic hypercalciuria (IH) is defined as that clinical situation in which an increase in urinary calcium excretion is observed, in the absence of hypercalcemia and other known causes of hypercalciuria. In recent years, its diagnosis in pediatric age has been more frequent because it has been known that it can debut with very different symptoms, in the absence of kidney stone formation. The discovery of genetic hypercalciuric stone-forming rats has allowed us to glimpse the pathophysiological mechanism of IH since they show many data in common with humans with IH as normal levels of blood calcium, intestinal calcium hyperabsorption, increased bone resorption and a defect in the renal tubular calcium reabsorption. In 1993, it was shown that in these animals there is an increase in the number of vitamin D receptors (VDR) in the intestine, which favors an increase in the functional capacity of calcitriol-VDR complexes that explains the increase in intestinal transport of calcium. The same happens at the bone level producing a greater resorption. In our opinion, IH is a "metabolic anomaly" or, better, an inheritable constitutive metabolic characteristic. In this sense, what patients with IH would inherit is the availability of having a greater number of VDRs in their cells than those with normal urinary calcium excretion. IH cannot be considered a sensu stricto disease, so pharmacological treatment must be individualized.
La hipercalciuria idiopática (HI) se define como aquella situación clínica en la que se comprueba un incremento en la eliminación urinaria de calcio, en ausencia de hipercalcemia y de otras causas conocidas de hipercalciuria. En los últimos años, su diagnóstico en la edad pediátrica ha sido más frecuente debido a que se ha conocido que puede debutar con síntomas muy diversos, en ausencia de formación de cálculos renales. El descubrimiento de las ratas hipercalciuricas ha permitido vislumbrar el mecanismo fisiopatológico de la HI ya que muestran muchos datos en común con los humanos con HI como niveles normales de calcemia, hiperabsorción intestinal de calcio, incremento de la resorción ósea y un defecto en la reabsorción tubular renal de calcio. En 1993, se demostró que en esos animales existe un incremento en el número de receptores de la vitamina D (VDR) del intestino, lo que favorece un aumento de la capacidad funcional de los complejos calcitriol-VDR que explica el incremento en el transporte intestinal de calcio. Lo mismo ocurre a nivel óseo produciéndose una mayor resorción. En nuestra opinión, la HI es una “anomalía metabólica” o, mejor, una característica metabólica constitutiva heredable. En este sentido, lo que los pacientes con HI heredarían es la disponibilidad de tener en sus células un mayor número de VDR que aquellas personas con calciurias normales. La HI no se puede considerar una enfermedad sensu stricto por lo que el tratamiento farmacológico debe ser individualizado.
Idiopathic hypercalciuria (IH) is defined as that clinical situation in which an increase in the urinary elimination of calcium is verified, in the absence of hypercalcemia and other known causes of hypercalciuria. It is the most frequent cause of renal lithiasis in both pediatric and adult age (about 40 % in series of children and 60 % in those of adults). IH is one of the most frequent metabolic abnormalities in humans, so the prevalence rates in healthy populations have been reported, between 0.6 and 12.5 %, depending on the country. In Spain, it is described a prevalence rate ranging from 3.8 and 7.8 %.
The association between excessive urinary calcium excretion and stone formation was described by Flocks in 1940.1 In 1953, Albright et al. used the term "idiopathic hypercalciuria" for the first time.2 Although initially it was believed that IH did not exist in children, at the end of the 1950s, Zetterström and Rosenkranz presented several pediatric cases in which lithiasis and IH were present in the same patient. In 1962, Valverde published the first Spanish pediatric cases, some of them with concomitant urinary tract infection (UTI).3 The same year, two pediatric groups based in Paris led by Royer and Gentil, respectively, reported 6 cases of children with nanism, osteopenia or rickets, renal abnormalities (proteinuria, polyuria) and hypercalciuria. For years, this variety was known as "Royer type IH".4 In the last decades no new cases have been described, which suggests that those first patients were carriers of other well-defined tubulopathies at the present time, such as "hypophosphatemic rickets with hypercalciuria", "familial hypomagnesemia with hypercalciuria" or "Dent's disease ».
Clinic. Prelitiasis conceptInitially, IH was associated only with renal colic and expulsion of stones. In recent years, its diagnosis has been much more frequent than in the past because it has been known that it can begin in the pediatric age with very different symptoms and in the absence of kidney stone formation. Therefore, at the beginning of the 80s, a new concept was introduced that nowadays is evident. We refer to the ability to diagnose in childhood those subjects who could suffer from nephritic colic years later, especially in adulthood. This situation, almost typical of the pediatric age, we have called "prelitiasis",5,6 although the term has not had much acceptance. In this line, in 1981, three different pediatric groups published that macro- or microscopic hematuria could be a manifestation of IH and, therefore, be suggestive of the possibility to suffer from nephritic colic years7–9 years later. Moore, the author of the third of these manuscripts, pointed out that children affected with IH could start, apart from lithiasis, with other symptoms or signs such as dysuria, sterile leukocyturia, nocturnal enuresis, polaquiuria, urinary urgency and even discrete proteinuria.9 Shortly after, the association between UTI and IH10 would be published.
Subsequently, the relationship between microhematuria or macroscopic hematuria and the presence of IH was confirmed in various studies, so that 25–42% of children referred to specialized centers to evaluate hematuria had IH. However, hematuria is not specific to IH since other metabolic abnormalities causing lithiasis, such as hyperuricosuria or hyperoxaluria, may be associated with hematuria. The production of experimental hypercalciuria for a short period of time is not accompanied by hematuria,11 so it is assumed that any metabolic abnormality that can cause microcrystals lithiasis, may produce a lesion of the renal tubular epithelium and secondary hematuria. In addition, it has been described the association of hypercalciuria, hyperuricosuria and nephrolithiasis with thin basal membranes nephropathy.12 The presence of recurrent abdominal pain "not typical of renal colic" has also been associated with IH.
Special mention deserves the association between IH and UTI. In 1987, Cervera et al. published that the frequency of UTI in his patients with IH was 48.9 %,10 which contrasts with the estimated prevalence of UTI in the general population, 1–2 % in boys and 3–5 % in girls.13 These initial data were subsequently confirmed in several studies.5,14 Similarly, it has been described that the frequency of hypercalciuria in series of children with UTI ranges between 20 %15 and 44 %.16 In a recent work published by our Group it was observed that the frequency of prelitiasis (including the calculation of the calcium/citrate ratio) in children with UTI by Escherichia coli was 47.5 % and there was a positive family history for urolithiasis in 68.3 % of cases17 ; in that article we postulated that children with prelitiasis must have, in some way, a constitutive reduced defense capacity against these bacteria. In this sense, the finding reported in the aforementioned study is very significant, in which a greater frequency of renal scars was observed in children with prelitiasis in relation to those with a normal urinary metabolic study.17
Pathogenic theories. Hypercalciuric ratsInitial studiesThe first studies on the pathophysiology of IH were published in 1965 by Edwards and Hodgkinson.18 These authors maintained their patients in a low calcium diet while administering a calcium chelating agent (EDTA) to prevent enteral absorption of alcium. Once the intestinal component was abolished, the calciuria was reduced, but when observing that it was not annulled, these authors concluded that the origin of that hypercalciuria should be exclusively renal. It is reasonable to think that this prolonged loss of urinary calcium of renal origin, in the case of being exclusive, should reduce ionic calcium and increase PTH levels. Well, when PTH could be determined, Pak et al., in 1974, observed that their levels were normal, so it was ruled out that IH was exclusively renal in origin.19 It is likely that the few patients with litiasis and IH who had hyperparathyroidism had to be those who had had their dairy products suppressed with the mistaken idea of reducing the episodes of lithiasis.20 In the aforementioned article by Pak et al., a test labeled with its name was disseminated that attempted to delimit adult patients with IH into two subtypes according to the underlying pathophysiological mechanism, absorptive or renal.19 In 1996, Aladjem et al. published that, at least in children, the behavior of calciuria was different 3–7 years after the first test and that the classification of patients in two different pathophysiological subtypes could be «artificial».21 Currently, that test is not used.
The discovery that some patients had elevated PTH levels prompted Alhava et al. to request bone densitometry in the distal radius of 75 patients with renal lithiasis.22 As one good example serendipity, they found that the patients had significantly lower values of bone mineral density compared to controls. Subsequent work confirmed these findings.23,24 Years later it was learned that the cause of osteopenia was not secondary hyperparathyroidism.
In the mid-1980s, when the levels of calcitriol [1,25- (OH)2 D3] could be determined, it was found that some patients showed high levels of calcitriol, these results brought back the intestinal theory of IH.25,26 In addition, at that time it was known that the hypercalciuria observed in cases with hyperproduction of prostaglandin E2 (PGE2) was reduced after treatment with indomethacin.27 Buck et al. treated 43 patients with IH with this prostaglandin inhibitor for 2–4 weeks and it was found a normalization of calciuria, which would implication of PGE2 in the origin of hypercalciuria.28 Shortly afterwards, it was suggested that the osteopenia observed in patients with IH could be secondary to bone resorptive effect of prostaglandin. 29 The Rodríguez-Iturbe Group, in Venezuela, showed that the urinary levels of PGE2 were increased in patients with IH suggesting that high PGE2 was a primary phenomenon in the etiology of IH.30
To further complicate this issue, in the 1980s it was published that the loss urinary phosphate could be the cause of the hypercalciuria observed in IH.31 It is known that in cases with marked hyperphosphaturia, hypophosphatemia can occur, which favors the synthesis of calcitriol which, in turn, increases intestinal calcium absorption with the consequence of hypercalciuria. However, the loss of phosphate observed in some cases of IH is not very intense and therefore insufficient to reduce phosphatemia. This mechanism, when intense, explains the hypercalciuia observed in phosphate deficit32 or in tubulopathy called «hereditary hypophosphatemia with hypercalciuria».33 So much of contrasting mechanisms had Martínez-Maldonado to state that the pathophysiology of IH34 was "continually changing."
Another possible etiological component is the diet. Although from the end of the 19th century it was known that urolithiasis has36 genetic bases, in the 80s and early 90s it was argued that excessive intake was the cause of IH. In 1982, two independent groups reported that excess sodium in the diet could be the cause of IH.37,38 Osmotic diuresis caused by the increase in filtered sodium would partially prevent renal tubular calcium reabsorption. In addition, Breslau et al. suggested that hypercalciuria induced by excess dietary sodium was accompanied by an increase in calcitriol synthesis.37 Additionally, in the late 1980s, it was published that IH could be secondary to an increase in protein intake.39 This excess could explain the reduction in bone mineral density.40 It was the return to the bone hypothesis of IH. In fact, with the excess of proteins of animal origin there is an acidic overload that need to be buffered in the bone, in addition to the other body buffers. Thus, Bataille et al. they observed in their patients a direct correlation between calciuria and urinary elimination of hydroxyproline.40 Both dietary theories of IH had to be confronted with some observations made in the 1990s by two groups of pediatric nephrologists. We refer to the fact that children diagnosed with IH, who had not had time to perpetrate excessive diets, already showed a reduction in bone mineral density at diagnosis.41,42
The increased production of cytokines by monocytes. Integration of the findings so far describedSince the early 1970s, it was known the existence of a factor other than PTH known as the "osteoclast activating factor" that has resorptive effect.43 This factor could be produced by some of the blood leukocytes.44 Pacifici et al. showed that blood monocytes isolated from patients with IH produced increased amounts of cytokine, such as interleukin-1 〈 (IL-1), the colony stimulating factorof granulocyte-macrophage and tumor necrosis factor 〈(TNF - 〈).45 An increased activity of these cytokines could reduce bone mineral density of patients with IH. The Pacifici Group's finding was subsequently confirmed.46,47
Weisinger, in 1996, postulated a pathophysiological theory that combined the different findings previously published on the pathophysiology of IH.48 IL-1 and the other cytokines would stimulate bone resorption45–47 and, secondarily, the production of PGE249 which, in turn, would induce the synthesis of calcitriol.50 It is known that calcitriol has a paradoxical effect on the bone in such a way that, in high amounts, it also stimulates its resorption.51,52 Hypercalciuria would be caused, therefore, by an increase in bone resorption and by an increase in intestinal calcium absorption due to the effect of calcitriol. In addition a diet rich in salt37,38 or in animal protein39,40 would further increase the calciuria.
Using a hyposaline overload, our Group described the existence of a discrete distal loss of salt in some adult patients with IH.53 It is known that inflammatory mediators such as IL-1 and TNF reduce sodium epithelial transport because, as indicated, they induce an increase in the synthesis of PGE 249,54 and they reduce the expression of the epithelial sodium channel (ENaC) and / or Na + -K + -ATPase of the basolateral membrane.55 This renal loss of sodium would also increase calciuria, so in some patients with IH, the calciuria could have a triple origin: bone, intestinal and renal.
Despite this wide range of pathophysiological paths, still the reason why monocytes are stimulated to increase cytokine production was unknown.
Did hypercalciuric rats provided the answer to the pathophysiology of idiopathic hypercalciuria?In 1979, the existence of rats with spontaneous hypercalciuria56 (genetic hypercalciuric stone-forming [GHS]) was described. Rat cross mating, lead to a successive increase in calciuria in the following generations.57 In search of the mechanism of hypercalciuria, Bushinsky and Favus observed that the rats of the fourth generation had a marked urinary elimination of calcium due to an increase in intestinal calcium absorption, although the levels of calcitriol were normal.57 When the rats were subjected to a reduced calcium diet, a decrease in calciuria was observed but it did not normalized, suggesting that the increase in intestinal calcium absorption was a mechanism that explained, at least in part, the hypercalciuria observed in these animals.58 In 1993, it was shown that in these rats there was an increase in the number of intestinal vitamin D receptors (VDR),59 which favored an increase in the functional capacity of the calcitriol-VDR complexes that explained, therefore, the increase in intestinal calcium transport. In addition, Yao et al. they found that, in these animals, there was an increased response of the VDRs to minimal doses of calcitriol, which implies that with normal amounts of calcitriol were enough to increase calciuria.60 As we have indicated above, when the rats were subjected to a low calcium diet, the urinary calcium excretion was reduced, but this was still higher than the dietary intake, which suggested, in addition, another pathogenic component. Indeed, Krieger et al. they showed that this increased sensitivity to calcitriol was also expressed in the bones of these animals, inducing greater bone resorption,61 which seemed to demonstrate that the bone also plays a role in the development of hypercalciuria. Thus, Bushinsky et al. demonstrated in these animals under a low- calcium diet that alendronate bisphosphonate reduced calciuria to a level below that would correspond to dietary intake.62
Later, it was found that in hypercalciuric rats there is also a defect in renal tubular reabsorption of calcium.63 This fact has been attributed the activation of the calcium receptor (CaR) that would suppress the activity of the calcium sensitive potassium channel (ROMK) in the ascending limb of the loop of Henle.64 By decreasing the electrical gradient in the tubular lumen due to the reduced concentration of potassium, calcium absorption by the paracellular route would be reduced, as occurs in Bartter's syndrome. This mechanism is the one that has been involved in "familial hypercalciuric hypocalcemia," caused by mutations in function gain in the gene encoding the calcium-sensitive receptor. This entity is sometimes accompanied by hypokalemia (Bartter syndrome type 5).65
In summary, hypercalciuric rats have many features in common with humans with IH, namely, normal calcemia, intestinal calcium hyperabsorption, increased bone resorption and a defect in renal tubular reabsorption of calcium. In addition, their calcitriol levels are normal,64 in the same way as most patients with HI.
In 2004, Favus et al. showed that peripheral monocytes of humans with IH have an increase in the number of VDR,66 that is, the same as previously observed in hypercalciuric rats.59 However, it should be remembered that a few years earlier Zerwekh et al. did not observe differences in the concentration of VDR of skin fibroblasts from subjects with absorptive IH, as compared with a control group.67
Some gene polymorphisms involvedIn recent years, it has been described that patients with hypercalciuric lithiasis may be carriers of certain polymorphisms in some genes that encode certain proteins involved in tubular calcium or phosphate reabsorption (VDR, SLC34A1, SLC34A4, CLDN14, CaS R, TRPV6), or, in the prevention of precipitation of calcium salts (CaSR, MGP, OPN, PLAU, UMOD).68–70
Fig. 1 summarizes and integrates all the findings involved in the pathophysiology of IH. Note, therefore, that in some cases of IH both mechanisms could be added, namely an increase in the functional capacity of the calcitriol-VDR complexes and the existence of gene polymorphisms that favor the formation of stones. It remains to be determined whether the hyperactivity of the cytokine-producing monocytes described in the IH (previous section) is also related to an increase in VDR in these cells.
Hypercalciuria and islanders. The case of the island of La Gomera. An inmunologic form?
In the 90s we found that in the pediatric population of the island of La Gomera (Canary Islands) there was a very high frequency of hypercalciuria, one of the highest in the world (16 % vs. 3.8 % in the control group). The island morphology is abrupt and radially furrowed by canyons that arise from the center of the island, which makes communication very difficult between the different areas of the island. The prevalence in different populations of the island was analyzed and it was found to be higher in the most geographically isolated part of the island (Valle Gran Rey, 28.4 %; Chipude-La Dama, 19.6 %) with respect to Vallehermoso (14, 9 %) and Hermigua (13.7 %), closer to San Sebastián (10.6 %), the island capital.71 It was observed that the risk of suffering hypercalciuria among children who had the 4 grandparents from the island (16.6 %) was 2.85 times higher than those who did not have any grandparents from the island (7.3 %). When sibling groups were studied, the presence of hypercalciuria was found in 50 % of them.71 This higher frequency of hypercalciuria in the most remote areas and, therefore with more limited communicated could be related to a higher frequency of inbreeding than in the better communicated populations.
In the island of La Gomera it has been proven, until the beginning of this century, an inbreeding index (marriages between first cousins) that is estimated to be a 25.8 %.72 The pathophysiological hypothesis of our Group that could explain our findings was published in 2006.73 In the island, for the past centuries, there has been a very poor sanitary, economic and nutritional conditions. It is reasonable that only the best-nourished children and those who allegedly defended themselves better from certain infections reached adulthood. It is therefore probable that those that we could define as immunological "strong" would have more options to reach adulthood. Our hypothesis is that the children of La Gomera were with IH would be descendants of those subjects immunologically stronger. Consequently, insularity and the associated high rate of consanguinity would not be the cause but rather the way to perpetuate these favorable immune conditions of the subjects that survived.73 Was this favorable capacity the result of an excess of VDR favored by consanguinity, similar to that described in hypercalciuric rats?59 Is IH the expression of a immunological phenotype? It should not be forgotten that vitamin D is a modulator of the immune system.74,75 Was an increase in the functional capacity of calcitriol-VDR complexes beneficial to facilitate the survival of these children? However, this condition of superior immunological aptitude would not be applicable to all types of infections since, as indicated above, the frequency of UTI is much higher in children with IH than in the control population.5,10,14–17 The above concepts can hide epigenetic mechanisms, ie modifications in gene expression that are not due to altered DNA sequence and establish a relationship between genetic and environmental influences that determine a phenotype.
In this regard, it has been described a high incidence of urolithiasis and/or hypercalciuria in other island populations such as the Fiji Islands,76–78 Puerto Rico79 or Iceland.80 Rudan et al. analyzed the susceptibility to suffer from nephrolithiasis in the inhabitants of 3 Croatian islands. The mean inbreeding coefficient (F) of each population was estimated. The prevalence of kidney stones was 1.5 % in the group of towns with a low F coefficient, 2.3 % in the group with a moderate F coefficient and 5.4 % in the group with a high F coefficient.81
It remains to be known, if in places where inbreeding was common in the past, it is more frequent to have certain lithiasis-promoting polymorphisms. In the case of La Gomera, the polymorphism found is located in the SLC34A3 gene (9q33.2–34.2) that encodes the phosphate transporter NPT2c and is responsible for 15 % of the proximal phosphate reabsorption.68,70 Mutations in this gene give rise to the picture called «hypophosphatemic rickets with hypercalcuria ».33 This finding would explain, for example, the renal loss of phosphate described in some patients with IH.31 In the case of Iceland, the polymorphism described is located in the CLDN14 gene that is involved in the tubular reabsorption of calcium in the ascending the loop of Henle.69
Bone mineral density associated hypocitraturiaThe osteopenia of IH patients with lithiasis was described by Alhava et al. in 1976,22 and confirmed by other authors is in the 80s23,24; it is related to an increase in bone resorption linked to a greater production of cytokines,45–47 PGE229 and/or calcitriol.51,52 The increased bonr resorption is the result of a greater sensitivity to calcitriol, as described in hypercalciuric rats.61
In the few IH cases with bone biopsies available, it has been observed an increase in osteoclastic activity.82 However, in some series, it has been described a reduction of osteoblastic activity,23 which has been attributed to infrequent phosphate deficit83 or to some degree of "functional hypoparathyroidism" (low remodeling) in some of these patients with serum calcium levels close to the high normal limit.84
Heilberg et al. studied bone biopsies performed in 36 adult patients with IH.85 As compared with controls, it was observed a high expression of the Receptor Activator for Nuclear Factor k B Ligand (NF-ĸ B) (RANKL), suggesting that the increase in bone resorption present in patients with IH is mediated by this peptide. The expression of IL-1 and basic fibroblast growth factor (bFGF) was similar to that of the controls. Based on these results, these authors consider that the high cytokine expression previously described in patients with IH may not be the cause loss of bone mineral density. From these results, Heilberg et al. consider that the pathophysiologic basis of IH would be an increased expression of VDRs, which, by increasing the functional capacity of the calcitriol-VDR complexes, would enhance the intestinal absorption of calcium, and stimulate the bone expression RANKL.85
In the pediatric field, our Group described for the first time that the Z value of bone mineral density (Z-BMD) was less than −1 in 30.1 % of children studied with IH.42 In a subsequent study, we found the existence of a Z-BMD value less than −1 in the lumbar spine in 42.5 % of a group of girls and in 47.5 % of their mothers (lumbar spine and/or femoral neck) that shared the increase in calciuria.86
Our Group quantified bone markers to confirm an excessive bone resorptive activity in pediatric patients with IH. In children with normal bone mineral density a direct correlation was observed between levels of osteocalcin (bone formation marker a) and those of tartrate-resistant acid phosphatase (resorption marker). Such a relationship did not exist in osteopenic children.42
In a subsequent work, we evaluated two more sensitive resorption markers, deoxypyridinoline (DPir) and the C-terminal telopeptide fraction of urine collagen (CrossLaps or CTx). Children with IH, with or without osteopenia, showed significantly higher values of the DPir/Cr and CrossLaps/Cr ratios than the controls. In contrast, osteocalcin levels were only significantly higher in those patients who had normal bone mineral density (Fig. 2). These data would confirm that in children with IH there is an increase in osteoclastic activity and only those with an adequate compensatory osteoblastic response would have a normal bone mineral density.87
 in hypercalciuric children with normal bone mineral density and osteopenia, in relation to controls.87")
Mean values of osteocalcin and deoxypyridinoline (D-Pir/Cr) in hypercalciuric children with normal bone mineral density and osteopenia, in relation to controls.87
Finally, it is difficult to explain why some patients with IH throughout the course of the disease, usually in adolescence, normalize calciuria21 and reduce citraturia.88–90 In our clinical practice we have also observed that in some of the families of children with IH, some of their parents with urolithiasis have hypocitraturia and normocalciuria. In two longitudinal studies, we have observed that in many patients, both in adolescence91 and in adulthood,90 there is spontaneous catch-up of bone mineral density. Alkali compounds are necessary to form bone. Let´s not forget that bone is the largest reserve of alkaline salts in the body.92,93 Our hypothesis is that the late hypocitraturia observed in IH would be an indirect sign of the existence of a greater osteoblastic activity that would require, in addition to higher amounts of calcium (normalization of calcium) and phosphate, the competition for supplementary amounts of alkalis. It is known that filtered citrate is reabsorbed in the proximal tubule to produce new bicarbonate through the action carbonic anhydrase, both luminal and intracellular. In summary, the proximal reabsorption of citrate would be increased if bone activity is increased. This would be an explanation for the late hypocitraturia observed in some of these patients.
Genetic considerations metabolic abnormality or disease?In 1874, Clubbe sent a letter to the editor of The Lancet in which he mentioned that he had attended a family in which 5 of its members had suffered urinary stones.35 After almost one century, Resnick et al. described a higher frequency of urolithiasis in the 625 parents and siblings of 106 subjects prone to calcium oxalate stone formation, compared to 576 relatives of the spouses of the probands. The authors ruled out a monogenic inheritance and formulated the hypothesis that the tendency to form calcium oxalate kidney stones is regulated by a polygenic system.94 In 1979, Coe et al. they studied the relatives of 9 patients with IH who had calcium oxalate stones and verified the existence of IH in 26 of the 76 relatives studied, without differences in age and sex; in addition, 43 % of first-degree relatives hadIH. The authors suggested that it was an autosomal dominant inheritance.95 Subsequently, several papers insisted on the polygenic genetic character of IH.36,96–98 Indeed, studies aimed at looking for a single causal gene of IH have been unsuccessful both with respect to unknown genes99–101 and to other already known genes related in some way to calcium metabolism.102–104
In our opinion, IH is a "metabolic abnormality" or, better, a metabolic characteristic that is constitutive and it is inherited as the color of the skin, the number of fingers or the final height. In this sense, what patients with IH would inherit is the availability of having in their cells a greater number of VDR than individuals with normal calciurias. For this reason, a sensu strict it cannot be considered a disease although it may predispose in some cases to the formation of kidney stones, the appearance of urinary infections and the development of osteoporosis at long-term. In addition, many people transmit this condition to their offspring but are asymptomatic, do not form calculi nor they have a reduced bone mineral density. This is the fundamental reason why the use of drug treatment should be carefully selected.
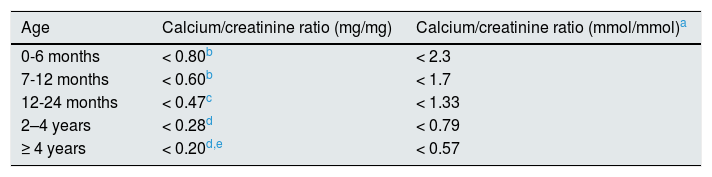
Diagnosis, monitoring and treatmentA calciuria greater than 4mg/kg/day in a 24h urine collection is the accepted criterion for the diagnosis of hypercalciuria. To confirm the diagnosis this determination must be repeated or quantify the calcium/creatinine ratio in an isolated urine sample (Table 1). In a family hypercalciuric members and/or renal lithiasis, blood determinations may be obviated or delayed since usually they will are normal. However, to confirm that a hypercalciuria is an IH, levels of calcemia, intact PTH, ions (including chloride) and acid base balance must be normal. In the follow-up, and to simplify the management of patients with IH, it has been postulated to determine calcium, citrate and creatinine in isolated urine samples at two different times of the day, namely before dinner and in the first morning urine, so it would not be necessary to collect urine from 24h.105 If calcium / citrate ratio in either urine exceeds 0.33mg/mg there is a risk of urinary crystallization.106,107 With this method, it has been shown that urinary concentrations of calcium and the calcium/citrate ratio are modified throughout the day so that the urine formed during the night is the most lithogenic.105 Sometimes in adolescence, there sis s simultaneous reduction of calciuria and citraturia,88–90 so the lithogenic risk may be less than 0.33 given the reduction in calciuria, despite hypocitraturia.
Normal values of the calcium/creatinine ratio according to age.
| Age | Calcium/creatinine ratio (mg/mg) | Calcium/creatinine ratio (mmol/mmol)a |
|---|---|---|
| 0-6 months | < 0.80b | < 2.3 |
| 7-12 months | < 0.60b | < 1.7 |
| 12-24 months | < 0.47c | < 1.33 |
| 2–4 years | < 0.28d | < 0.79 |
| ≥ 4 years | < 0.20d,e | < 0.57 |
Kruse et al. (Eur J Pediatr. 1984; 143: 25–31) and Melián et al.71 Urine not collected after fasting.
Since, as previously reasoned, IH is a metabolic abnormality rather than a disease, dietary treatment should initially be encouraged by stimulating the intake of water, fruits (especially citrus fruits), vegetables, blue fish (due to its rich in acids ⌉-3 fatty) and whole grains (due to its richness in phytate). In addition, the abuse of salt and protein should be avoided. Pharmacological treatment should be reserved for two circumstances; first, clinical data of sustained dysuria, frequent macroscopic hematuria, or recurrent symptoms of kidney stones. Secondly, if stones or nephrocalcinosis are observed by ultrasound. Even in the presence of microcalculi, dietary treatment could be indicated, at least for one year, to observe the evolution. Although therapeutic details are beyond the scope of this review, a marketed product based on phytate (Broken®) and 3 types of drugs are available, namely thiazides, potassium citrate and bisphosphonates.
Special comment deserves bone mineral density. As indicated before, both spontaneous recovery of bone mineral density occurs both in adolescence91 and in adulthood.90 In a longitudinal study, our Group observed that, at least in children, the improvement of bone mineral density was more related to the increase in body mass than to the use of thiazides.108 Presently, performing bone densitometry in children with IH seems especially indicated in cases of fractures or marked bone pain. This is the opposed of what should be promoted in the elderly with IH and/or urolithiasis.
Financial supportSome of the works referred to in this review were funded by the Ministry of Education, Culture and Sports of the Canary Islands Government (BOC of the Concession Resolution: No. 110 [08/17/93]), HE. Island Council of La Gomera (Resolution date: 12/29/94) and by the Health Research Fund (FIS) (Project number: 98/1179).
Conflict of interestsThe authors declare no conflict of interest.
Please cite this article as: García Nieto VM, Yanes MIL, Carreño PT, Suarez GP, Mesa TM. La hipercalciuria idiopática revisada. ¿Anomalía metabólicao enfermedad? Nefrologia. 2019;39:592–602.



