






La hipercalciuria idiopática (HI) se define como aquella situación clínica en la que se comprueba un incremento en la eliminación urinaria de calcio, en ausencia de hipercalcemia y de otras causas conocidas de hipercalciuria. En los últimos años, su diagnóstico en la edad pediátrica ha sido más frecuente debido a que se ha conocido que puede comenzar con síntomas muy diversos, en ausencia de formación de cálculos renales. El descubrimiento de las ratas hipercalciúricas ha permitido vislumbrar el mecanismo fisiopatológico de la HI ya que muestran muchos datos en común con los humanos con HI, como niveles normales de calcemia, hiperabsorción intestinal de calcio, incremento de la resorción ósea y un defecto en la reabsorción tubular renal de calcio. En 1993, se demostró que en esos animales existe un incremento en el número de receptores de la vitamina D (VDR) del intestino, lo que favorece un aumento de la capacidad funcional de los complejos calcitriol-VDR que explica el incremento en el transporte intestinal de calcio. Lo mismo ocurre a nivel óseo produciéndose una mayor resorción. En nuestra opinión, la HI es una «anomalía metabólica» o, mejor, una característica metabólica constitutiva heredable. En este sentido, lo que los pacientes con HI heredarían es la disponibilidad de tener en sus células un mayor número de VDR que aquellas personas con calciurias normales. La HI no se puede considerar una enfermedad sensu stricto, por lo que el tratamiento farmacológico debe ser individualizado.
Idiopathic hypercalciuria (IH) is defined as that clinical situation in which an increase in urinary calcium excretion is observed, in the absence of hypercalcemia and other known causes of hypercalciuria. In recent years, its diagnosis in pediatric age has been more frequent because it has been known that it can debut with very different symptoms, in the absence of kidney stone formation. The discovery of genetic hypercalciuric stone-forming rats has allowed us to glimpse the pathophysiological mechanism of IH since they show many data in common with humans with IH as normal levels of blood calcium, intestinal calcium hyperabsorption, increased bone resorption and a defect in the renal tubular calcium reabsorption. In 1993, it was shown that in these animals there is an increase in the number of vitamin D receptors (VDR) in the intestine, which favors an increase in the functional capacity of calcitriol-VDR complexes that explains the increase in intestinal transport of calcium. The same happens at the bone level producing a greater resorption. In our opinion, IH is a ‘metabolic anomaly’ or, better, an inheritable constitutive metabolic characteristic. In this sense, what patients with IH would inherit is the availability of having a greater number of VDRs in their cells than those with normal urinary calcium excretion. IH cannot be considered a sensu stricto disease, so pharmacological treatment must be individualized.
La hipercalciuria idiopática (HI) se define como aquella situación clínica en la que se comprueba un incremento en la eliminación urinaria de calcio, en ausencia de hipercalcemia y de otras causas conocidas de hipercalciuria. Es la causa más frecuente de litiasis renal tanto en la edad pediátrica como en la adulta (alrededor del 40% en series de niños y del 60% en las de adultos). La HI es una de las anomalías metabólicas más frecuentes en el ser humano, de tal modo que se han referido tasas de prevalencia en población sana, según los países, de entre 0,6 y 12,5%. En España, las tasas de prevalencia descritas oscilan entre 3,8 y 7,8%.
La asociación entre una excesiva excreción urinaria de calcio y la formación de cálculos fue descrita por Flocks en 19401. En 1953, Albright et al. emplearon por primera vez el término «hipercalciuria idiopática»2. Aunque inicialmente se creía que la HI no existía en niños, a finales de la década de los 50, Zetterström y Rosenkranz presentaron varios casos pediátricos en los que coincidían litiasis e HI. En 1962, Valverde publicó los primeros casos pediátricos españoles, algunos de ellos con infección de vías urinarias (IVU) concomitante3. Ese año, dos grupos pediátricos radicados en París dirigidos por Royer y Gentil, respectivamente, comunicaron 6 casos de niños con nanismo, osteopenia o raquitismo, alteración renal (proteinuria, poliuria) e hipercalciuria. Durante años, esa variedad fue conocida con el nombre de «HI tipo Royer»4. En las últimas décadas no se han descrito casos nuevos, lo que hace pensar que aquellos primeros pacientes eran portadores de otras tubulopatías bien definidas en el momento actual, como «raquitismo hipofosfatémico con hipercalciuria», «hipomagnesemia familiar con hipercalciuria» o «enfermedad de Dent».
Clínica. Concepto de prelitiasisInicialmente, la HI solo se asoció con cólicos nefríticos y expulsión de cálculos. En los últimos años, su diagnóstico ha sido mucho más frecuente que antaño debido a que se ha conocido que puede comenzar en la edad pediátrica con síntomas muy diversos, en ausencia de formación de cálculos renales. Por ello, a principios de los años 80, se introdujo un nuevo concepto, hoy en día evidente. Nos referimos a la capacidad de poder diagnosticar en la infancia a aquellos sujetos que podrían padecer cólicos nefríticos años después, especialmente, en la edad adulta. A esta situación, casi propia de la edad pediátrica, la hemos denominado «prelitiasis»5,6, aunque el término no ha tenido mucha fortuna. En este sentido, en 1981, tres grupos pediátricos diferentes publicaron que la hematuria macro- o microscópica podía ser una manifestación de HI y, por tanto, ser un dato indicativo de la capacidad potencial de padecer cólicos nefríticos años más tarde7-9. El autor del tercero de estos trabajos, Moore, apuntó que los niños afectos de HI podían empezar, aparte de con litiasis, con otros síntomas o signos como disuria, leucocituria estéril, enuresis nocturna, polaquiuria, urgencia miccional e, incluso, proteinuria discreta9. Poco después, se publicaría la asociación entre IVU e HI10.
Posteriormente, se confirmó en diversos trabajos la relación entre microhematuria o hematuria macroscópica y la presencia de HI, de tal modo que del 25 al 42% de los niños remitidos a centros de referencia por hematuria tienen HI. No obstante, la hematuria no es específica de la HI puesto que otras anomalías metabólicas causantes de litiasis, como hiperuricosuria o hiperoxaluria, pueden asociarse con hematuria. La producción de hipercalciuria experimental durante corto tiempo no se acompaña de hematuria11, por lo que se supone que cualquier anomalía metabólica causante de litiasis puede producir microcristales, una lesión del epitelio tubular renal y hematuria secundaria. Además, se ha descrito la asociación de hipercalciuria, hiperuricosuria y nefrolitiasis con nefropatía de las membranas basales finas12. La presencia de dolor abdominal recurrente «no típico de cólico renal» se ha asociado, asimismo, con HI.
Mención especial merece la asociación entre HI e IVU. En 1987, Cervera et al. publicaron que la frecuencia de IVU en sus pacientes con HI era del 48,9%10, lo que contrasta con la prevalencia estimada de IVU en población general que es del orden del 1-2% en varones y del 3-5% en niñas13. Esos datos iniciales fueron confirmados posteriormente en varios trabajos5,14. Del mismo modo y a la inversa, se ha descrito que la frecuencia de hipercalciuria en series de niños con IVU oscila entre el 20%15 y el 44%16. En un trabajo reciente publicado por nuestro Grupo hemos observado que la frecuencia de prelitiasis (incluyendo el cálculo del cociente calcio/citrato) en niños con IVU por Escherichia coli era del 47,5% y existía una historia familiar positiva para urolitiasis en el 68,3% de los casos17. En ese artículo postulamos que los niños con prelitiasis deben tener, de alguna forma, una capacidad defensiva constitutiva reducida frente a esas bacterias. En este sentido, es muy significativo el hallazgo comunicado en el mencionado trabajo en el que se observó una mayor frecuencia de cicatrices renales en los niños con prelitiasis en relación con aquellos con un estudio metabólico urinario normal17.
Teorías patogénicas. Las ratas hipercalciúricasLos estudios inicialesLos primeros estudios sobre la fisiopatología de la HI fueron publicados en 1965 por Edwards y Hodgkinson18. Estos autores sometieron a sus pacientes a una dieta pobre en calcio al tiempo que les administraban un quelante de la absorción enteral de ese ion (EDTA). Una vez abolido el componente intestinal, la calciuria se redujo, pero al observar que no se anulaba, esos autores concluyeron que el origen de la hipercalciuria debía ser exclusivamente renal. Es razonable pensar que esa pérdida prolongada de calcio urinario de origen renal, en el caso de ser exclusiva, debería reducir el calcio iónico e incrementar los niveles de PTH. Pues bien, cuando se pudo determinar la PTH, Pak et al., en 1974, observaron que sus niveles eran normales, por lo que se descartaba que la HI fuera de origen exclusivamente renal19. Es probable que los pocos pacientes con litiasis e HI que tenían hiperparatiroidismo debían ser aquellos a los que se les había suprimido los lácteos de la dieta con la errónea idea de reducir los episodios de litiasis20. En el citado artículo de Pak et al., se difundió una prueba etiquetada con su nombre que intentaba deslindar a pacientes adultos con HI en dos subtipos según el mecanismo fisiopatológico subyacente, absortivo o renal19. En 1996, Aladjem et al. publicaron que, al menos en niños, el comportamiento de la calciuria difería de 3 a 7 años después de la primera prueba y que la clasificación de los pacientes en dos subtipos fisiopatológicos diferentes podía ser «artificial»21. En la actualidad, esa prueba no se utiliza.
El descubrimiento de que algunos pacientes tenían niveles elevados de PTH impulsó a Alhava et al. a solicitar densitometrías óseas en la porción distal del radio a 75 pacientes con urolitiasis22. Dentro del mejor ejemplo de serendipia, encontraron que los pacientes tenían valores significativamente inferiores de densidad mineral ósea con respecto a los controles. Trabajos posteriores confirmaron sus hallazgos23,24. Años después se supo que la causa de la osteopenia no era el hiperparatiroidismo secundario.
A mediados de los años 80, cuando se pudieron determinar los niveles de calcitriol [1,25-(OH)2D3], se comprobó que algunos pacientes mostraban niveles elevados, con lo que hubo un retorno a la teoría intestinal de la HI25,26. Además, en ese momento se sabía que la hipercalciuria observada en los casos con hiperproducción de prostaglandina E2 (PGE2) se reducía tras instaurarse tratamiento con indometacina27. En ese sentido, Buck et al. trataron a 43 pacientes con HI con ese inhibidor de las prostaglandinas durante 2-4 semanas y comprobaron la normalización de la calciuria, lo que podía indicar una implicación de PGE2 en su origen28. Poco después, se sugirió que la osteopenia observada en los pacientes con HI podía ser secundaria a un efecto resortivo prostaglandin-dependiente29. El Grupo de Rodríguez-Iturbe, en Venezuela, demostró que los niveles urinarios de PGE2 estaban incrementados en pacientes con HI. Estos autores sugirieron que se trataba de un fenómeno primario en su etiología30.
Para complicar aún más la cuestión, en los años 80 se publicó que la pérdida urinaria de fosfato podía ser la causa de la hipercalciuria observada en la HI31. Es conocido que en los casos en los que la hiperfosfaturia es importante, se puede producir hipofosfatemia, lo que favorece la síntesis de calcitriol que, a su vez, aumenta la absorción intestinal de calcio con la consecuencia de hipercalciuria. No obstante, la pérdida de fosfato observada en algunos casos de HI es poco intensa y, por tanto, insuficiente para reducir la fosfatemia. Ese mecanismo, cuando es intenso, explica la hipercalciuria observada en el déficit de fosfato32 o en la tubulopatía denominada «hipofosfatemia hereditaria con hipercalciuria»33. Tanto mecanismo dispar dio lugar a que Martínez-Maldonado manifestara que se «estaba cambiando continuamente» la fisiopatología de la HI34.
Aún queda por recordar otro posible importante componente etiológico, el dietético. A pesar de que desde finales del siglo XIX35 se sabía que la urolitiasis tiene bases genéticas36, en los años 80 y principios de los 90 se defendió que los excesos en la ingesta eran la causa de la HI. En 1982, dos grupos independientes notificaron que el exceso de sodio de la dieta podía ser la causa de la HI37,38. La diuresis osmótica producida por el incremento de sodio filtrado impediría parcialmente la reabsorción tubular renal de calcio. Además, Breslau et al. sugirieron que la hipercalciuria inducida por el exceso de sodio dietético se acompañaba de un incremento de la síntesis de calcitriol37. Por otra parte, a finales de los 80, se publicó que la HI debía ser secundaria a un incremento en la ingesta de proteínas de la dieta39. Ese exceso podía explicar la reducción de la densidad mineral ósea40. Se trataba del retorno a la hipótesis ósea de la HI. En efecto, con el exceso de proteínas de origen animal se produce una sobrecarga ácida que precisa tamponarse en el hueso, además de en los otros tampones corporales. Así, Bataille et al. observaron en sus pacientes una correlación directa entre la calciuria y la eliminación urinaria de hidroxiprolina40. Ambas teorías dietéticas de la HI se enfrentaban con algunas observaciones realizadas en los años 90 por dos grupos de nefrólogos pediátricos. Nos referimos a que los niños diagnosticados de HI, que no habían tenido tiempo de cometer excesos en la dieta, ya mostraban una reducción de la densidad mineral ósea al diagnóstico41,42.
La producción incrementada de citocinas por parte de los monocitos. Integración de los hallazgos descritos hasta ese momentoDesde principios de los años 70 se conocía la existencia de un factor distinto de la PTH denominado «factor activador de los osteoclastos» con un efecto resortivo óseo43. Este factor podía ser producido por algunos de los leucocitos sanguíneos44. Pacifici et al. demostraron que los monocitos sanguíneos aislados de pacientes con HI producían una cantidad incrementada de citocinas, tales como la interleucina-1α (IL-1), el factor estimulante de colonias de los granulocitos-macrófagos y el factor de necrosis tumoral-α (TNF-α)45. Un incremento de la actividad de estas citocinas tendría la capacidad de reducir la densidad mineral ósea de los pacientes con HI. El hallazgo del Grupo de Pacifici fue confirmado posteriormente46,47.
Weisinger, en 1996, postuló una teoría fisiopatológica que aunaba los diferentes hallazgos anteriormente publicados sobre la fisiopatología de la HI48. La IL-1 y las otras citocinas estimularían la resorción ósea45-47 y, secundariamente, la producción de PGE249 que, a su vez, induciría la síntesis de calcitriol50. Es conocido que el calcitriol tiene un efecto paradójico sobre el hueso de tal modo que, en cantidades elevadas, estimula, también, su resorción51,52. La hipercalciuria sería ocasionada, por tanto, por un incremento de resorción ósea y por un aumento de la absorción intestinal de calcio debido al efecto del calcitriol. Junto con ello, una dieta rica en sal37,38 o en proteínas de origen animal39,40 acrecentaría, aún más, la calciuria.
Por otra parte, mediante sobrecarga hiposalina, nuestro Grupo describió la existencia de una discreta pérdida salina distal en algunos pacientes adultos con HI53. Se sabe que los mediadores inflamatorios como IL-1 y TNF reducen el transporte epitelial de sodio gracias a que, como se ha indicado, inducen un incremento de la síntesis de PGE249,54 y a que reducen la expresión del canal epitelial de sodio (ENaC) y/o de la Na+-K+-ATPasa de la membrana basolateral55. Esta pérdida renal de sodio, incrementaría, asimismo, la calciuria por lo que, en algunos pacientes con HI, la calciuria podría tener un triple origen: óseo, intestinal y renal.
A pesar de este amplio abanico fisiopatológico, no se conocía la razón por la que los monocitos se estimulaban para incrementar la producción de citocinas.
¿Las ratas hipercalciúricas han aportado la solución a la fisiopatología de la hipercalciuria idiopática?En 1979, se describió la existencia de ratas con hipercalciuria espontánea56 (genetic hypercalciuric stone-forming [GHS]). Al cruzarse entre ellas se comprobó un sucesivo incremento de la calciuria en las siguientes generaciones57. En busca del mecanismo de la hipercalciuria, Bushinsky y Favus observaron que las ratas de la cuarta generación tenían una marcada eliminación urinaria de calcio gracias a la existencia de un incremento de la absorción intestinal de calcio, si bien los niveles de calcitriol eran normales57. Cuando se sometieron las ratas a una dieta reducida en calcio, se comprobó un descenso de la calciuria aunque sin normalizarse, lo que sugería que el incremento en la absorción intestinal de calcio era un mecanismo que explicaba, al menos en parte, la hipercalciuria observada en estos animales58. En 1993, se demostró que en estos existía un incremento en el número de receptores de la vitamina D (VDR) del intestino59, lo que favorecía un aumento de la capacidad funcional de los complejos calcitriol-VDR que explicaba, por ende, el incremento en el transporte intestinal de calcio previamente descrito. Además, Yao et al. comprobaron que, en estos animales, existía una respuesta incrementada de los VDR a mínimas dosis de calcitriol, lo que implica que no eran necesarios niveles muy elevados del mismo para amplificar su respuesta e incrementar sobremanera la calciuria60. Como hemos indicado más arriba, cuando las ratas eran sometidas a una dieta baja en calcio, se reducía la excreción urinaria de calcio, pero esta aún era superior a la ingesta dietética, lo que sugería, además, otro componente patogénico. En efecto, Krieger et al. mostraron que ese incremento de la sensibilidad al calcitriol se expresaba, asimismo, en los huesos de esos animales induciendo una mayor resorción ósea61, con lo que parecía demostrarse que el hueso juega, asimismo, un papel en el desarrollo de la hipercalciuria. De este modo, Bushinsky et al. demostraron en estos animales sometidos a una dieta baja en calcio, que el bisfosfonato alendronato reducía la calciuria a un nivel por debajo del correspondiente a la ingesta dietética62.
Más tarde, se comprobó que en las ratas hipercalciúricas existe, asimismo, un defecto en la reabsorción tubular renal de calcio63. Este hecho se ha atribuido a una activación del receptor sensible a calcio (CaR) que suprimiría la actividad del canal de potasio sensible a calcio (ROMK) en la rama ascendente del asa de Henle64. Al menguar el gradiente eléctrico en la luz tubular dada la menor concentración de potasio, se reduciría la absorción de calcio por la vía paracelular, como ocurre en el síndrome de Bartter. Este mecanismo es el que se ha involucrado en la «hipocalcemia hipercalciúrica familiar», causada por mutaciones de ganancia de función en el gen que codifica el receptor sensible a calcio. Esta entidad se acompaña, en ocasiones, de hipopotasemia (síndrome de Bartter tipo 5)65.
En resumen, las ratas hipercalciúricas tienen muchos datos en común con los humanos con HI, a saber, niveles normales de calcemia, hiperabsorción intestinal de calcio, incremento de la resorción ósea y un defecto en la reabsorción tubular renal de calcio. Además, sus niveles de calcitriol son normales64, del mismo modo que la mayoría de los pacientes con HI.
En 2004, Favus et al. demostraron que los monocitos periféricos de los humanos con HI tienen un incremento del número de VDR66, es decir, lo mismo que se había observado previamente en las ratas hipercalciúricas59. No obstante, es preciso recordar que unos años antes Zerwekh et al. no observaron diferencias en la concentración de VDR, al estudiar fibroblastos de la piel de sujetos con HI absortiva, en relación con el grupo control67.
Algunos polimorfismos génicos implicadosEn los últimos años, se ha descrito que los pacientes con litiasis hipercalciúrica pueden ser portadores de ciertos polimorfismos en algunos genes que codifican determinadas proteínas involucradas en la reabsorción tubular de calcio o fosfato (VDR, SLC34A1, SLC34A4, CLDN14, CaSR, TRPV6), o bien, en la prevención de la precipitación de sales de calcio (CaSR, MGP, OPN, PLAU, UMOD)68-70.
En la figura 1 se resumen e integran todos los hallazgos implicados en la fisiopatología de la HI que han sido enumerados. Obsérvese, por tanto, como en algunos casos de HI podrían sumarse ambos mecanismos, a saber, un incremento de la capacidad funcional de los complejos calcitriol-VDR y la existencia de polimorfismos génicos favorecedores de la formación de cálculos. Queda por determinar si la hiperactividad de los monocitos productores de citocinas descrita en la HI (apartado anterior) está igualmente relacionada con un incremento de VDR en esas células.
Hipercalciuria e insularidad. El caso de la isla de La Gomera. ¿Una forma de ser inmunológica?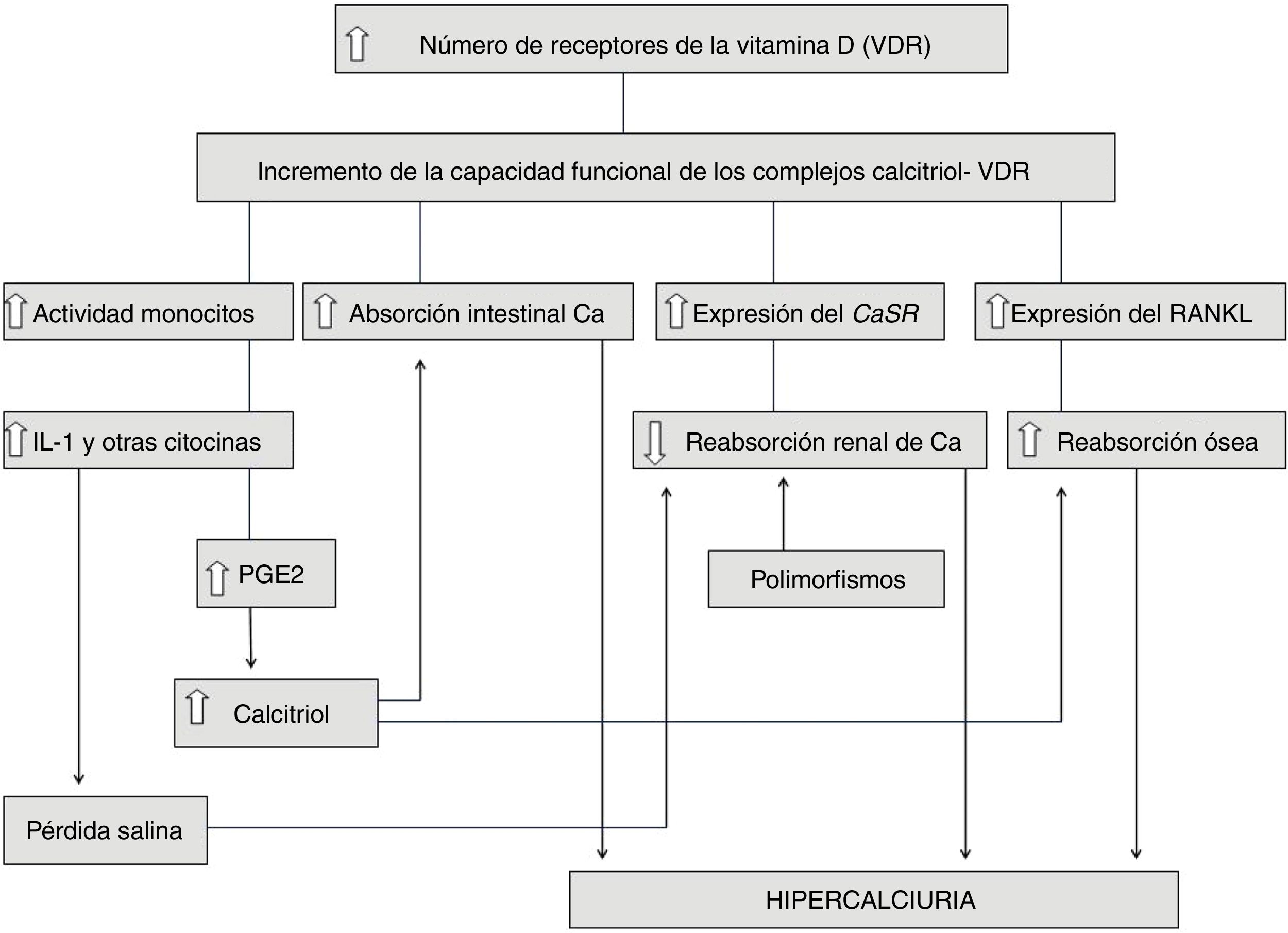
En la década de los 90 comprobamos que en la población pediátrica de la isla de La Gomera (Islas Canarias) existía una frecuencia muy elevada de hipercalciuria, una de las más altas del mundo (16% frente al 3,8% en el grupo control). La morfología insular es abrupta y surcada radialmente por barrancos que surgen desde el centro de la isla, con lo que es difícil la comunicación entre las distintas zonas de la misma. Se analizó la prevalencia en distintas poblaciones de la isla y se comprobó que era más elevada en las más aisladas geográficamente (Valle Gran Rey, 28,4%; Chipude-La Dama, 19,6%) con respecto a Vallehermoso (14,9%) y Hermigua (13,7%), más cercanas a San Sebastián (10,6%), capital insular71. Se observó que el riesgo de padecer hipercalciuria entre los niños que tenían los 4 abuelos originarios de la isla (16,6%) era 2,85 veces superior al de aquellos que no tenían ningún abuelo procedente de la misma (7,3%). Cuando se estudiaron grupos de hermanos se comprobó la presencia de hipercalciuria en el 50% de los mismos71. Esta mayor frecuencia de hipercalciuria en las zonas más alejadas y, por ende, peor comunicadas podría tener relación con una mayor frecuencia de endogamia que en las poblaciones mejor comunicadas.
En la isla de La Gomera se ha comprobado, hasta principios de este siglo, un índice endogámico (matrimonios entre primos hermanos) que se estima en un 25,8%72. La hipótesis fisiopatológica de nuestro Grupo que pudiera explicar nuestros hallazgos fue publicada en 200673. En el pasado y durante siglos, en la isla existieron unas condiciones sanitarias, económicas y nutricionales muy deficientes. Es razonable que solo llegaran a la edad adulta los niños mejor nutridos y los que, presuntamente, se defendían mejor de ciertas infecciones de las que están actualmente protegidos gracias a los antibióticos y a las vacunas. Es, pues, probable que tuvieran más opciones de alcanzar la edad adulta los que podríamos definir como «fuertes» inmunológicos. Nuestra hipótesis es que los niños de La Gomera con HI serían descendientes de esos sujetos más aptos desde el punto de vista inmunológico. Por consiguiente, la insularidad y la alta tasa de consanguinidad asociada, no serían la causa sino la forma de perpetuar esas condiciones inmunológicas favorecedoras de los sujetos que sobrevivían73. ¿Esa aptitud era debida a un exceso de VDR favorecido por la consanguinidad, similar a lo descrito en las ratas hipercalciúricas59? ¿Es la HI la expresión de una forma de ser inmunológica? No debe olvidarse que la vitamina D es un modulador del sistema inmune74,75. ¿Un incremento de la capacidad funcional de los complejos calcitriol-VDR era beneficioso para facilitar la supervivencia de esos niños? No obstante, esa condición de superior aptitud inmunológica no sería aplicable a todo tipo de infecciones puesto que, como se ha indicado antes, la frecuencia de IVU es mucho más elevada en niños con HI que en la población control5,10,14-17. Los conceptos mencionados pueden esconder mecanismos epigenéticos, es decir, modificaciones en la expresión de genes que no obedecen a una alteración de la secuencia del ADN y que establecen una relación entre las influencias genéticas y ambientales que determinan un fenotipo.
En relación con el tema, se ha descrito una alta incidencia de urolitiasis y/o hipercalciuria en otras poblaciones insulares como las islas Fidji76-78, Puerto Rico79 o Islandia80. Rudan et al. investigaron la susceptibilidad para padecer nefrolitiasis en los pobladores de 3 islas croatas. Se estimó el coeficiente medio de endogamia (F) de cada población. La prevalencia de cálculos renales era del 1,5% en el grupo de pueblos con un coeficiente F bajo, del 2,3% en el grupo con un coeficiente F moderado y del 5,4% en el grupo con un coeficiente F elevado81.
Falta por saber, especialmente, si en lugares en los que en el pasado fue habitual la consanguinidad son más frecuentes, además, ciertos polimorfismos favorecedores de litiasis, como se ha indicado previamente. En el caso de La Gomera, el polimorfismo encontrado está ubicado en el gen SLC34A3 (9q33.2–34.2) que codifica el transportador de fosfato NPT2c y que es responsable del 15% de la reabsorción proximal de fosfato68,70. Mutaciones en este gen originan el cuadro denominado «raquitismo hipofosfatémico con hipercalciuria»33. Este dato explicaría, por ejemplo, la pérdida renal de fosfato descrita en algunos pacientes con HI31. En el caso de Islandia, el polimorfismo descrito está ubicado en el gen CLDN14 que está involucrado en la reabsorción tubular de calcio en la rama ascendente del asa de Henle69.
La densidad mineral ósea. La hipocitraturia asociadaEn párrafos precedentes se ha indicado que la osteopenia propia de los pacientes litiásicos con HI fue descrita por Alhava et al. en 197622, confirmada por otros autores en los años 8023,24 y relacionada con un incremento de la resorción ósea ligada a una mayor producción de citocinas45-47, PGE229 y/o calcitriol51,52. Esta acción resortiva incrementada estaría relacionada con una mayor sensibilidad al calcitriol, como se ha descrito en las ratas hipercalciúricas61.
En los pocos casos de HI en los que se han realizado biopsias óseas se observó un incremento de la actividad osteoclástica82. No obstante, en algunas series, se ha descrito una reducción de la actividad osteoblástica23, que se ha atribuido al infrecuente déficit de fosfato83 o a un cierto grado de «hipoparatiroidismo funcional» en algunos de estos pacientes (bajo remodelado), al exhibir niveles de calcemia cerca del límite alto de la normalidad84.
Heilberg et al. estudiaron muestras procedentes de biopsias óseas realizadas a 36 pacientes adultos con HI85. Observaron, en relación con los controles, una alta expresión del ligando de unión al receptor activador del factor nuclear kappaB (NF-κB) (RANKL), lo que sugiere que el incremento en la resorción ósea presente en los pacientes con HI sería mediado por este péptido. La expresión de IL-1 y del factor de crecimiento fibroblástico básico (bFGF) fue similar a la de los controles. En razón a esos resultados, esos autores consideran que la alta expresión de citocinas descrita previamente en pacientes con HI podría no tener una relación causal en la pérdida de densidad mineral ósea previamente descrita. A partir de estos resultados, Heilberg et al. consideran que lo primario en la fisiopatología de la HI sería el incremento ya descrito de los VDR que, al favorecer un aumento de la capacidad funcional de los complejos calcitriol-VDR, además de incrementar la absorción intestinal de calcio, podría estimular la expresión ósea de RANKL85.
En el ámbito pediátrico, nuestro Grupo describió por primera vez que el valor Z de la densidad mineral ósea (Z-DMO) era menor de −1 en el 30,1% de los niños estudiados con HI42. En un estudio posterior, comprobamos la existencia de un valor de Z-DMO menor de −1 en la columna lumbar en el 42,5% de un grupo de niñas y en el 47,5% de sus madres (columna lumbar y/o cuello femoral) con las que compartían el incremento en la calciuria86.
Nuestro Grupo cuantificó, asimismo, algunos marcadores óseos para intentar confirmar el mecanismo resortivo en los pacientes pediátricos con HI. Así, en los niños con densidad mineral ósea normal se observó una correlación directa entre los niveles de osteocalcina (marcador de formación ósea) y los de fosfatasa ácida tartrato-resistente (marcador de resorción). Esta relación desaparecía en los niños osteopénicos42.
En un trabajo posterior, estudiamos dos marcadores de resorción más sensibles, la desoxipiridinolina (DPir) y la fracción telopeptídica C-terminal del colágeno en orina (CrossLaps o CTx). Los niños con HI, con o sin osteopenia, mostraron valores significativamente más elevados de los cocientes DPir/Cr y CrossLaps/Cr que los controles. En cambio, los niveles de osteocalcina solo fueron significativamente más elevados en aquellos pacientes que tenían una densidad mineral ósea normal (fig. 2). Estos datos confirmarían que en los niños con HI existe un incremento de la actividad osteoclástica y que aquellos con densidad mineral ósea normal serían aquellos con una respuesta osteoblástica adecuada compensatoria87.
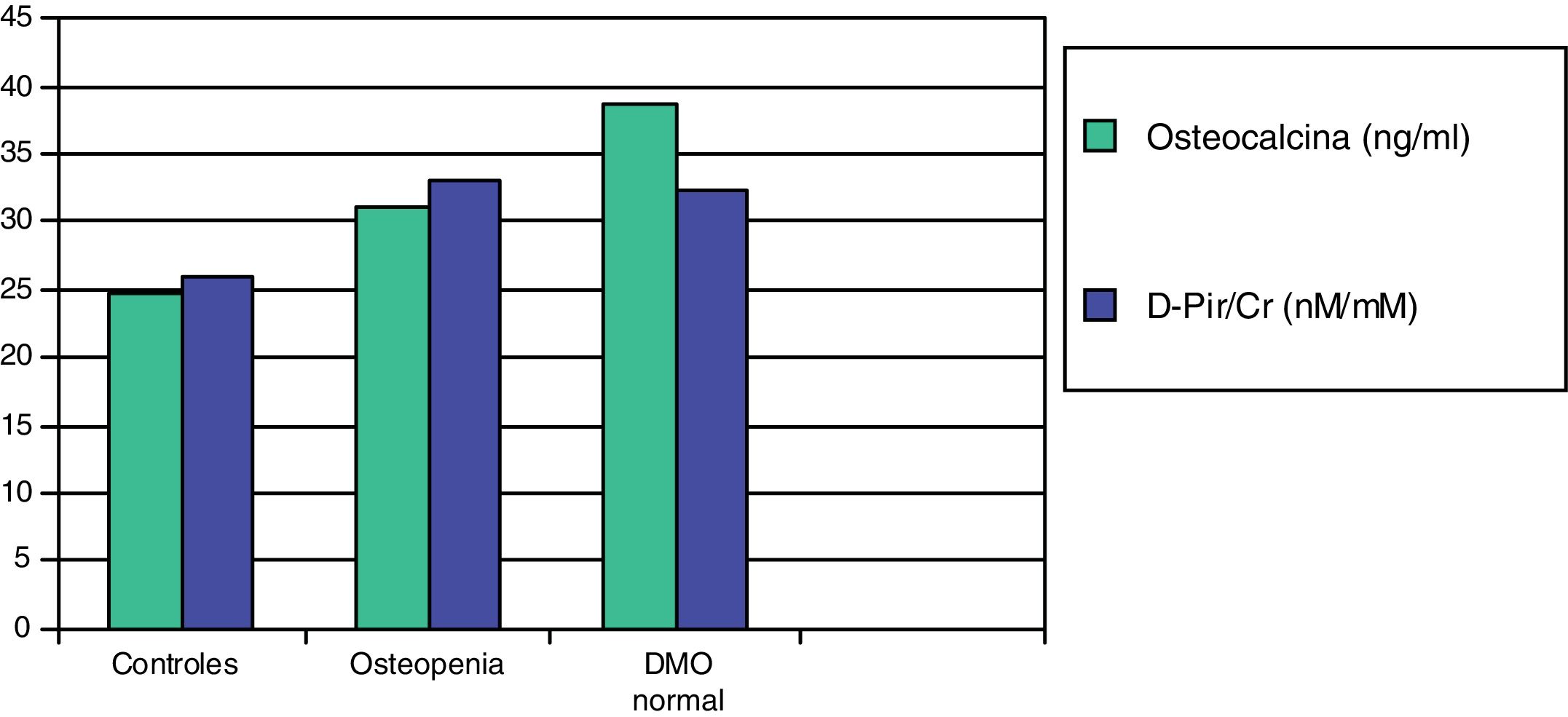
Valores medios de osteocalcina y de desoxipiridinolina (D-Pir/Cr) en niños hipercalciúricos con densidad mineral ósea normal y con osteopenia, en relación con los controles87.
Finalmente, es difícil explicar la razón por la que algunos pacientes con HI a lo largo de la evolución, generalmente en la adolescencia, normalizan la calciuria21 y reducen la citraturia88-90. En nuestra práctica clínica hemos observado, además, que en algunas de las familias de niños con HI, algunos de sus padres con urolitiasis tienen hipocitraturia y normolciuria. En dos estudios longitudinales, nuestro Grupo ha observado que en muchos pacientes, tanto en la adolescencia91 como en la edad adulta90, se produce una cierta recuperación (catch-up) espontánea de la densidad mineral ósea. Para formar hueso es necesario el concurso de álcalis. No debe olvidarse que el hueso es la mayor reserva de sales alcalinas del organismo92,93. Nuestra hipótesis es que la hipocitraturia tardía observada en la HI sería un signo indirecto de la existencia de una mayor actividad osteoblástica que precisaría, además de mayores cantidades de calcio (se normaliza la calciuria) y fosfato, el concurso de cantidades suplementarias de álcalis. Es conocido que el citrato filtrado se reabsorbe en el túbulo proximal para producir nuevo bicarbonato gracias a la existencia de la anhidrasa carbónica, tanto luminal como intracelular. En resumen, la reabsorción proximal de citrato estaría incrementada cuando existe una mayor actividad ósea. Podría ser esta una explicación para la hipocitraturia tardía observada en algunos de estos pacientes.
Consideraciones genéticas ¿Anomalía metabólica o enfermedad?En 1874, Clubbe dirigió una carta al editor de la revista The Lancet en la que mencionaba que había atendido una familia en la que 5 de sus miembros habían padecido cálculos urinarios35. Tuvo que pasar casi un siglo hasta que Resnick et al. describieron una mayor frecuencia de urolitiasis en los 625 padres y hermanos de 106 sujetos propensos a la formación de cálculos de oxalato de calcio, en comparación con los 576 familiares de los cónyuges de los probandos. Los autores descartaron una herencia monogénica y formularon la hipótesis de que la tendencia a formar cálculos renales de oxalato de calcio está regulada por un sistema poligénico94. En 1979, Coe et al. estudiaron los familiares de 9 pacientes con HI que habían eliminado cálculos de oxalato cálcico y comprobaron la existencia de HI en 26 de los 76 familiares estudiados, sin diferencias en la edad y el sexo; además, el 43% de los familiares en primer grado tenían HI. Los autores sugirieron que se trataba de una herencia autosómica dominante95. Posteriormente, varios trabajos insistieron en el carácter genético poligénico de la HI36,96-98. En efecto, los estudios dirigidos a buscar un único gen causal de la HI han sido infructuosos tanto con respecto a genes desconocidos99-101 como a los ya conocidos relacionados de alguna forma con el metabolismo del calcio102-104.
En nuestra opinión, la HI es una «anomalía metabólica» o, mejor, una característica metabólica constitutiva que se hereda como el color de la piel, el número de dedos de la mano o la talla final. En este sentido, lo que los pacientes con HI heredarían es la disponibilidad de tener en sus células un mayor número de VDR que aquellas personas con calciurias normales. Por esta razón, no se puede considerar una enfermedad sensu stricto aunque pueda predisponer en algunos casos a la formación de cálculos renales, a la aparición de infecciones urinarias y al desarrollo de osteoporosis a largo plazo. Además, muchas personas transmiten la condición a su descendencia pero son asintomáticas, no forman cálculos ni tienen reducción de la densidad mineral ósea. Esta es la razón fundamental por la que el uso de tratamiento farmacológico debe ser seleccionado.
Diagnóstico, seguimiento y tratamientoUna calciuria en orina de 24h superior a 4mg/kg/día en pacientes continentes es el criterio aceptado para el diagnóstico de hipercalciuria. Para confirmarlo debe repetirse esa determinación o cuantificar el cociente calcio/creatinina en orina aislada (tabla 1). En un ambiente familiar de hipercalciuria y/o litiasis renal pueden obviarse o retrasarse las determinaciones sanguíneas puesto que, habitualmente, serán normales. No obstante, para confirmar que una hipercalciuria se trata de una HI, deben ser normales los niveles de calcemia, PTH intacta, iones (incluido el cloro) y equilibrio ácido base. En el seguimiento, para simplificar el manejo de los pacientes con HI se ha postulado determinar calcio, citrato y creatinina en orina aislada en dos momentos del día, a saber, antes de la cena y en la primera orina de la mañana, por lo que no sería necesario recoger orina de 24h105. Si el cociente calcio/citrato en cualquiera de las dos orinas es superior a 0,33mg/mg existe riesgo de cristalización urinaria106,107. Con esa metodología se ha demostrado que las concentraciones urinarias de calcio y del cociente calcio/citrato se modifican a lo largo del día de tal modo que las orinas formadas durante la noche son las más litógenas105. En la adolescencia ocurre, en ocasiones, que se reducen al mismo tiempo la calciuria y la citraturia88-90, con lo que el riesgo litógeno puede ser menor de 0,33 dada la reducción de la calciuria, a pesar de la hipocitraturia.
Puesto que, como hemos razonado previamente, la HI es una anomalía metabólica y no una enfermedad, debe insistirse inicialmente en el tratamiento dietético estimulando la ingesta de agua, frutas (sobre todo, cítricos), verduras, pescado azul (por su riqueza en ácidos grasos ω-3) y cereales integrales (por su riqueza en fitato). Además, debe evitarse el abuso de sal y proteínas. El tratamiento farmacológico debe reservarse para dos circunstancias. En primer lugar, cuando existen datos clínicos marcados como disuria mantenida, hematuria macroscópica frecuente, o bien, cólicos nefríticos de repetición. En segundo lugar, cuando en la ecografía se observan cálculos o nefrocalcinosis. Incluso, ante la presencia de microcálculos, podría estar indicado el tratamiento dietético, al menos durante un año, para observar la evolución. Aunque los detalles terapéuticos quedan fuera del objetivo de esta revisión, se dispone de un producto comercializado a base de fitato (Broken®) y de 3 tipos de fármacos, a saber, tiazidas, citrato potásico y bisfosfonatos.
Comentario especial merece la densidad mineral ósea. Como hemos indicado antes, tanto en la adolescencia91 como en la edad adulta90 se produce una cierta recuperación espontánea de la densidad mineral ósea. En un estudio longitudinal, nuestro Grupo observó que, al menos en niños, la mejoría de la densidad mineral ósea estaba más relacionada con el incremento de la masa corporal que con el uso de tiazidas108. En el momento actual la realización de la densitometría ósea en niños con HI parece indicada, especialmente, en los casos de fracturas o de dolores óseos marcados. Esta postura es la opuesta a la que se debería fomentar en ancianos con HI y/o urolitiasis.
FinanciaciónAlgunos de los trabajos referidos en esta revisión fueron financiados por la Consejeria de Educación, Cultura y Deportes del Gobierno de Canarias (B.O.C. de la Resolución de concesión: n° 110 [17/08/93]), el Excmo. Cabildo Insular de La Gomera (fecha de la Resolución: 29/12/94) y por el Fondo de Investigación Sanitaria (FIS) (Número del proyecto: 98/1179).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



