




Sr. Director:
El síndrome antisintetasa (SAS) es una enfermedad poco frecuente englobada en las miopatías inflamatorias idiopáticas y que se caracteriza por la presencia de anticuerpos antisintetasa. La forma de presentación clínica del síndrome antisintetasa es variada y abarca polimiositis o dermatomiositis, poliartritis, enfermedad intersticial difusa pulmonar, fenómeno de Raynaud y lesiones cutáneas eritemato-violáceas hiperqueratósicas sobre zonas de articulación metacarpofalángicas e interfalángicas1,2. El SAS se debe a anticuerpos IgG dirigidos contra la enzima sintetasa. Se han identificado siete autoanticuerpos: anti-Jo1, anti-PL7, anti-PL12, anti-OJ, anti-EJ, anti-KS y anti-Wa, siendo el anti-Jo1 el mejor conocido.
La amiloidosis es una enfermedad del metabolismo de las proteínas que se caracteriza por el depósito extracelular de un conjunto de proteínas fibrilares en disposición beta plegada. Los tipos más importantes son la amiloidosis primaria o AL constituida fundamentalmente por fragmentos de cadenas ligeras de inmunoglobulinas y la amiloidosis secundaria o AA constituida por fibrillas de proteína A1-3. La afectación renal es frecuente en la amiloidosis secundaria, manifestándose con una gran variedad de signos y síntomas entre los que destacan proteinuria aislada, síndrome nefrótico, hipertensión arterial, hipotensión arterial, insuficiencia renal, etc. La amiloidosis secundaria a enfermedades reumáticas crónicas se ha convertido en el tipo más frecuente de amiloidosis secundaria.
Sólo se ha descrito en la bibliografía un caso con SAS y amiloidosis secundaria AA, pero este paciente presentaba un linfoma3-5.
CASO CLÍNICO
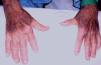
Presentamos el caso de un paciente hombre de 72 años diagnosticado por el servicio de reumatología de SAS anti-Jo1 positivo sin afectación miosítica con deterioro de la función renal. Se trataba de un paciente con antecedentes personales de hipertensión arterial, neuropatía intersticial, insuficiencia mitral moderada e hipertrofia ventricular izquierda con birrefringencia de septo. En un ingreso previo en el servicio de cardiología se realizó biopsia de grasa subcutánea con la sospecha de amiloidosis, que resultó negativa. En el ingreso actual ha sido derivado para el estudio de insuficiencia renal con creatinina de 2,9 mg/dl, proteinuria de 1,25 mg/24 h, sin otras alteraciones bioquímicas. En la exploración física llamaban la atención las telangiectasias en los párpados y las lesiones cutáneas eritemato-violáceas hiperqueratósicas (signo de Gottron) sobre las articulaciones metacarpofalángicas e interfalángicas («manos de mecánico», figura 1). El resto de la exploración física fue normal. Se completaron el estudio de autoinmunidad (ANA; ANCA negativo; C3 y C4 normales), inmunofijación en sangre y orina y proteinograma, sin alteraciones evidentes.
Se realizó radiografía de tórax en la que se evidenciaron cardiomegalia a expensas de aurículas y engrosamiento cisural con pinzamiento costofrénico derecho. En la ecografía abdominal visualizamos un riñón derecho de 9,2 cm con cortical ecogénica y un pequeño quiste simple de 1 cm en el polo inferior. El riñón izquierdo era de 9,3 cm, con características similares. En el polo superior existía un quiste simple de unos 3 cm, y en el inferior otro de 1,6 cm, con una tabicación calcificada en su interior.
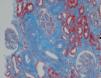
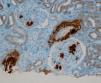
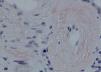
Se decidió realización de biopsia renal para filiar el diagnóstico, observándose 9 glomérulos esclerosados. Se detectaron, además, depósitos amorfos en glomérulo, vaso e intersticio, rojo Congo positivo y amiloide AA (IHQ) + en distribución glomérulo-intersticial, nodular y predominantemente perivascular. El diagnóstico anatomopatológico fue amiloidosis renal de tipo AA con afectación glomerular, intersticial y fundamentalmente vascular. (figura 2, figura 3, figura 4 y figura 5).
DISCUSIÓN
Se trata de un SAS sin datos clínicos ni analíticos de enfermedad muscular. Es un SAS sin afectación miosítica al que se asocia un deterioro de la función renal. La amiloidosis secundaria a enfermedades reumáticas crónicas se ha convertido en el tipo más frecuente de amiloidosis secundaria por el depósito de proteína betaamiloide, aunque sólo existe un caso en asociación con el SAS y además éste presentaba un linfoma.
En la bibliografía hemos encontrado miopatías inflamatorias relacionadas con la amiloidosis, como la amiloidosis por cuerpos de inclusión, sin embargo, en el caso del SAS, esta asociación no está descrita.
El tratamiento de la amiloidosis renal fue sintomático. El paciente ha sido seguido por el servicio de reumatología y ha recibido tratamiento con metotrexato, risedronato y suplementos con calcio y vitamina D, con buena respuesta y sin necesidad de tratamiento inmunosupresor5-7.
Al igual que se recomienda en otras enfermedades inflamatorias de larga evolución, creemos necesario plantear la realización de biopsia renal en pacientes con síndrome antisintetasa anti-Jo1 positivo y deterioro inexplicado de la función renal.
El paciente ha sido diagnosticado de insuficiencia renal crónica por amiloidosis secundaria AA con síndrome antisintetasa anti-Jo1 positivo sin afectación miosítica.

Figura 1. Manos de mecánico. Características del síndrome antisintetasa.

Figura 2. Técnica de hematoxilina-eosina en la que se observan nódulos amorfos en glomérulos y vasos.

Figura 3. Tricrómico de Masson en el que se observa una intensa fibrosis.

Figura 4. Estudio de inmunohistoquímica con depósitos de la proteína AA en vasos y glomérulos.

Figura 5. Tinción de Rojo Congo en la que se observan los habituales depósitos de amiloide en glomérulos y vasos.



