





To the Editor,
Antisynthetase syndrome (AS) is a rare disease in the idiopathic inflammatory myopathy group and is characterised by the presence of antisynthetase antibodies. The clinical presentation of antisynthetase syndrome is varied and includes polymyositis or dermatomyositis, polyarthritis, diffuse interstitial lung disease, Raynaud's phenomenon and erythematous-violaceous hyperkeratotic skin lesions on metacarpophalangeal and interphalangeal joint areas.1,2 AS is due to IgG antibodies directed against the enzyme synthase. Seven autoantibodies have been identified: anti-Jo-1, anti-PL7, anti-PL12, anti-OJ, anti-EJ, anti-KS and anti-Wa, with anti-Jo-1 being the best known.
Amyloidosis is a protein metabolism disease characterised by extracellular deposition of fibrillar protein set in beta fold arrangement. The most important are primary amyloidosis (AL), consisting mainly of fragments of light chain of immunoglobulins, and secondary amyloidosis (AA), consisting of protein A1-3 fibrils. Renal involvement is common in secondary amyloidosis, with a wide variety of signs and symptoms: isolated proteinuria, nephrotic syndrome, hypertension, hypotension, renal failure, etc. Amyloidosis secondary to chronic rheumatic diseases are the most common type of secondary amyloidosis.
Only one case of AS and secondary AA amyloidosis has been reported in the literature, but this patient had a lymphoma.3-5
Case report
We report the case of a 72-year-old man diagnosed by the rheumatology department with AS antiJo-1 positive without myositic damage and with impaired renal function. This patient had a history of hypertension, interstitial neuropathy, moderate mitral regurgitation and left ventricular hypertrophy with septal birefringence. In a previous hospital stay, the cardiology department performed a subcutaneous fat biopsy after suspicion of amyloidosis, which was negative. In this current hospitalisation, he has been referred for the study of renal failure, with creatinine 2.9mg/dl, proteinuria 1.25mg/24h, and no other biochemical changes. A physical examination revealed the telangiectasias of the eyelids and erythematous-violaceous hyperkeratotic skin lesions (Gottron sign) on the metacarpophalangeal and interphalangeal joints (“mechanic’s hands”, Figure 1). The remaining physical examination was normal. The autoimmunity study was completed (ANA, negative ANCA, normal C3 and C4), blood and urine immunofixation and serum protein studies were also performed and there were no apparent abnormalities.
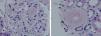
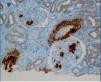
A chest x-ray was performed, which showed cardiomegaly at the expense of atria and fissural thickening with right costophrenic impingement. An abdominal ultrasound showed the right kidney to be 9.2cm with an echogenic cortical area and a small simple cyst of 1cm in the lower pole. The left kidney was 9.3cm, with similar characteristics. In the upper pole, there was a simple cyst of about 3cm, and one of 1.6cm in the lower pole, with calcified internal septation. It was decided to perform a renal biopsy for diagnosis, and 9 sclerotic glomeruli were seen. Also detected were amorphous deposits in the glomeruli, vessel and interstitial, Congo red-positive and AA amyloid + (IHC) in glomerular-interstitial, nodular and predominantly perivascular areas. The pathologic diagnosis was renal AA amyloidosis with glomerular, interstitial and mainly vascular affectation (Figures 2, 3, 4 and 5).
Discussion
This was an AS without clinical or analytical data of muscle disease or myositic affectation, which is associated with deterioration of renal function. Amyloidosis secondary to chronic rheumatic diseases are now the most common type of secondary amyloidosis caused by deposition of amyloid beta protein. However, there is only one case associated with AS, and that patient also had a lymphoma.
In the literature, we found inflammatory myopathies related with amyloidosis, such as amyloidosis due to inclusion bodies, however, this association is not described in the case of AS.
The treatment of renal amyloidosis was symptomatic. The patient was monitored by the rheumatology department, and treated with methotrexate, risedronate, and calcium and vitamin D supplements. He had a good response and did not need immunosuppressive therapy.5-7
As recommended for other long-duration inflammatory diseases, we believe it necessary to perform a renal biopsy in patients with anti-Jo-1 positive antisynthetase syndrome and an unexplained deterioration of renal function.
The patient was diagnosed with chronic renal failure due to amyloidosis secondary to AA with anti-Jo-1 positive antisynthetase syndrome without myositic affectation.

Figure 1. Mechanic's hands, characteristic of antisynthetase syndrome

Figure 2. Haematoxylin-eosin test showing amorphous nodules in glomeruli and vessels

Figure 3. Masson's trichrome showing intense fibrosis

Figure 4. Immunohistochemical study with AA protein deposits in vessels and glomeruli

Figure 5. Congo red stain showing the characteristic amyloid deposits in glomeruli and vessels



