




El término nefropatía tubulointersticial autosómica dominante (NTAD) engloba diferentes entidades causadas por alteraciones en los genes MUC1, UMOD, HNF1B, REN y más raramente SEC61A1, todas ellas con un patrón de herencia autosómico dominante1,2. Cursan con un deterioro progresivo de la función renal, sedimento urinario anodino, albuminuria normal o discretamente aumentada, ausencia de hipertensión arterial grave en estadios iniciales y ausencia de anomalías radiológicas o histológicas, lo que dificulta su diagnóstico3. Se estima que las NTAD suponen un 5% de las causas monogénicas de enfermedad renal crónica4.
La NTAD causada por una mutación en el gen UMOD que codifica la proteína uromodulina (NTAD-UMOD) es una de las 2 variantes más frecuentemente identificadas en la única cohorte española de NTAD publicada hasta la fecha5. La NTAD-UMOD supone el 1% de la enfermedad crónica avanzada que precisa tratamiento renal sustitutivo6. Aunque se ha propuesto la uromodulina urinaria como biomarcador para indicar el estudio genético de NTAD-UMOD, esta determinación no está disponible en la práctica clínica habitual7. Presentamos el caso de un adolescente con una mutación de novo del gen UMOD no descrita previamente.
Varón de 12 años que inicia el seguimiento en nuestro centro por nefropatía no filiada. Previamente era seguido en otro país por enfermedad renal crónica diagnosticada a los 3 años, sin que se le realizase biopsia renal ni otras pruebas de diagnóstico etiológico. Presenta a su llegada una creatinina sérica de 2,3mg/dl, filtrado glomerular por Schwartz actualizado 26ml/min/1,73 m2, hiperazoemia de 171,5mg/dl, hiperuricemia de 10,1mg/dl, acidosis metabólica (pH 7,36; pCO2 42mmHg, HCO3− 19,7 mmol/l), sin alteraciones iónicas. Parathormona de 486 pg/ml (15-77). La bioquímica urinaria muestra un déficit de concentración con una osmolalidad de 370 mOsm/kg H2O tras restricción hídrica, un cociente proteína/creatinina de 0,19mg/mg y sin hematuria ni otras alteraciones en el sedimento. Se observan en la ecografía riñones de tamaño en el límite inferior de la normalidad, hiperecogénicos, con mala diferenciación corticomedular y sin quistes. No presenta antecedentes personales reseñables ni antecedentes familiares de nefropatía o consanguinidad. Se descarta nefropatía en los padres y hermanos con función renal en sangre y orina normal, y ecografía renal sin alteraciones.
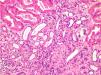
Se realiza una biopsia renal (figs. 1 y 2), compatible con daño tubulointersticial crónico moderado y con un patrón de inmunofluorescencia negativo a IgA, IgG, IgM, C1q, C3 y C4.


A lo largo de la evolución destaca una hiperuricemia de difícil control con una hipouricosuria marcada (en torno a 75mg/día/1,73 m2 [valores de referencia 344-850]). Se realiza el estudio del exoma filtrado por los genes relacionados a NTAD-UMOD y diagnósticos diferenciales, identificando una variante tipo missense en heterocigosis en el gen UMOD que ocasiona el cambio de la guanina en la posición 584 por una adenina (c.584G>A) en el exón 3 del tránscrito NM_003361.3. Esta modificación supone el cambio de un codón para cisteína por un codón para tirosina en la posición 195 (p.Cys195Tyr). Aunque esta variante no había sido descrita previamente en bases de datos poblacionales (GnomAD, ExAC, Proyecto 1000 genomas) y tampoco ha sido descrita como causante de enfermedad, 8 predictores in silico la catalogan como poco tolerada frente a 3 predicciones neutrales. Se ha descrito como patogénico el cambio de la cisteína en la posición 195 a fenilalanina. A pesar de no encontrarse en ningún dominio funcional de la proteína, otras variantes de cambio de sentido muy cercanas a esta variante han sido catalogadas como patogénicas8. Aplicando los criterios de interpretación de variantes publicados, concluimos que esta variante es probablemente patogénica y, por lo tanto, responsable del fenotipo presente en el paciente9. El estudio de segregación demuestra que se trata de una variante de novo.
El paciente evoluciona con deterioro de la función renal en menos de 2 años, precisando iniciar terapia renal sustitutiva mediante diálisis peritoneal a los 14 años. La progresión a enfermedad renal terminal es manifiestamente más rápida que la habitualmente descrita en otras mutaciones causantes de NTAD-UMOD, típicamente a partir de los 25 años10.
Concluimos que la mutación identificada de novo en este paciente es la causante de la NTAD-UMOD. Además, esta mutación podría condicionar una evolución más agresiva de la enfermedad, aunque serían necesarios estudios funcionales para demostrarlo, causando enfermedad renal terminal durante la adolescencia. Es posible que existan otros factores genéticos o ambientales que influyan en la progresión más agresiva de la tubulopatía en este paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.



