



The term autosomal dominant tubulointerstitial kidney disease (ADTKD) covers different conditions caused by abnormalities in the MUC1, UMOD, HNF1B, REN, and more rarely SEC61A1, genes, all with an autosomal dominant inheritance pattern.1,2 Initial findings are a progressive deterioration in kidney function, with unremarkable urine sediment, normal or slightly increased albuminuria, no severe hypertension in the initial stages and no radiological or histological abnormalities, making diagnosis difficult.3 It is estimated that ADTKD accounts for 5% of monogenic causes of chronic kidney disease.4
ADTKD, caused by a mutation in the UMOD gene, which encodes the uromodulin protein (ADTKD-UMOD), is one of the two variants most commonly identified in the only Spanish ADTKD cohort published to date.5 One percent of advanced chronic disease requiring renal replacement therapy is caused by ADTKD-UMOD.6 Although urinary uromodulin has been proposed as a biomarker to indicate the genetic study of ADTKD-UMOD, the test is not available in routine clinical practice.7 We present the case of an adolescent with a de novo mutation of the UMOD gene, which has not previously been reported.
This was a 12-year-old male who began follow-up at our centre for kidney disease of unknown cause. He had previously been followed up in a different country for chronic kidney disease diagnosed at three years of age. No renal biopsy or other aetiological diagnostic tests had been performed. On arrival, he had a serum creatinine of 2.3 mg/dl, revised Schwartz equation glomerular filtration rate of 26 ml/min/1.73 m2, azotaemia of 171.5 mg/dl, hyperuricaemia of 10.1 mg/dl, metabolic acidosis (pH 7.36; pCO2 42 mmHg, HCO3 − 19.7 mmol/l), with no electrolyte abnormalities. Parathyroid hormone 486 pg/mL (15–77). Urine biochemistry showed a concentration deficit with an osmolality of 370 mOsm/kg H2O after fluid restriction, a protein/creatinine ratio of 0.19 mg/mg, and no haematuria or other abnormalities in the sediment. Ultrasound showed kidneys at the lower limit of normal size, hyperechogenic, with poor corticomedullary differentiation and no cysts. The patient had no relevant medical history or family history of kidney disease or consanguinity. Kidney disease was ruled out in his parents and siblings; normal kidney function tests in blood and urine and no abnormalities on kidney ultrasound.
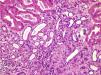
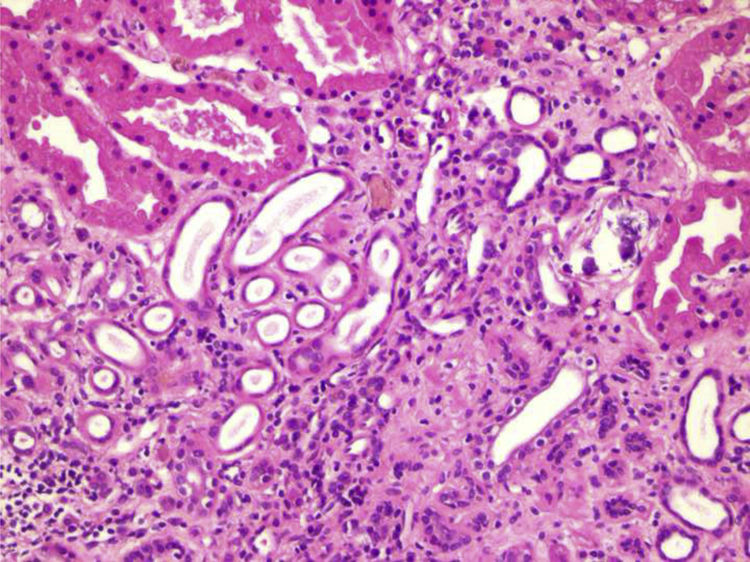
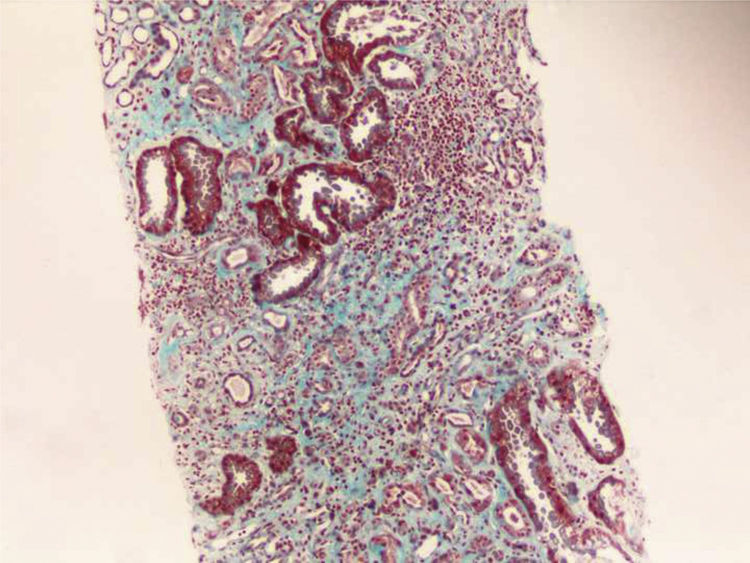
Renal biopsy was performed (Figs. 1 and 2) and analysis was consistent with moderate chronic tubulointerstitial damage and with a negative immunofluorescence pattern for IgA, IgG, IgM, C1q, C3 and C4.
The patient had difficult-to-control hyperuricaemia throughout, with marked hypouricosuria (around 75 mg/day/1.73 m2 [reference values 344–850]). Exome sequencing was performed, filtering for the genes related to ADTKD-UMOD and differential diagnoses, identifying a heterozygous missense type variant in the UMOD gene involving the change of guanine in position 584 to an adenine (c.584 G > A ) in exon 3 of the NM_003361.3 transcript. This modification involves the change of a cysteine codon to a tyrosine codon at position 195 (p.Cys195Tyr). Although this variant had not been previously described in population databases (GnomAD, ExAC, Project 1000 Genomes) and has not been described as causing disease, eight in silico predictors classify it as poorly tolerated compared to three neutral predictions. The change of cysteine at position 195 to phenylalanine has been described as pathogenic. Despite not being found in any functional domain of the protein, other missense variants very close to this variant have been classified as pathogenic.8 Applying the published criteria for interpretation of variants, we conclude that this variant is probably pathogenic and is therefore responsible for the phenotype present in our patient.9 The segregation study shows that it is a de novo variant.
The patient's kidney function continued to deteriorate, and less than two years later, at the age of 14, he had to begin renal replacement therapy with peritoneal dialysis. The progression to end-stage renal disease is much faster than usually described in other ADTKD-UMOD-causing mutations, where patients are typically over 25 before this happens.10
We conclude that the de novo mutation identified in this patient is the cause of his ADTKD-UMOD. In addition, this mutation may dictate a more aggressive disease course, causing end-stage renal disease during adolescence. However, functional studies would be necessary to prove this. Other genetic or environmental factors may influence the more aggressive progression of tubulopathy in this patient.
Conflicts of interestThe authors declare that they have no conflicts of interest.



