



La detección temprana de la nefropatía por enfermedad de Fabry es de interés, pues su tratamiento es más eficaz en estadios precoces. Ha sido estudiada por biomarcadores moleculares y tisulares, pero estos poseen desventajas que dificultan su uso rutinario. El propósito del presente trabajo es describir el rol del nefrólogo en el diagnóstico de la enfermedad y las variables clínicas asociadas a nefropatía en pacientes afectados.
Material y métodosEstudio transversal. Se incluyeron pacientes de tres centros de referencia de Argentina.
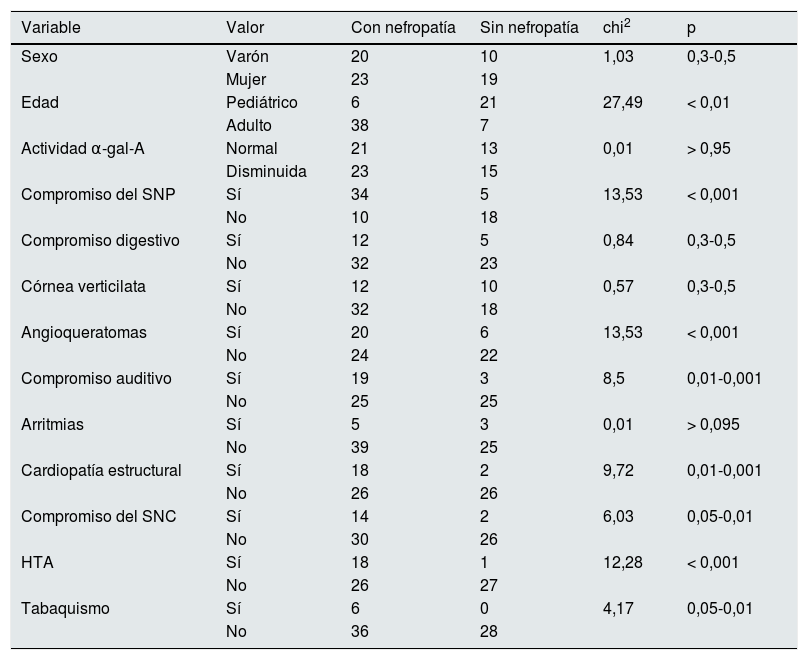
ResultadosSe estudiaron 72 pacientes (26,26±16,48años): 30 (41,6%) varones y 42 (58,4%) mujeres; 27 pediátricos y 45 adultos. Se detectaron 14 «casos índice», el 50% diagnosticados por nefrólogos. Se halló nefropatía en 44 pacientes (61%): 6 pediátricos y 38 adultos. Dos tipos de variables clínicas se asociaron a nefropatía: a)compromiso del sistema nervioso periférico (p≤0,001), angioqueratomas (p≤0,001) y compromiso auditivo (p=0,01-0,001), siendo estas manifestaciones clínicas tempranas del fenotipo más severo de la enfermedad, y b)cardiopatía estructural (p=0,01-0,001) y compromiso del sistema nervioso central (p=0,05-0,01), que son complicaciones mayores y tardías, responsables de la morbimortalidad aumentada y la menor expectativa de vida.
ConclusiónEl nefrólogo cumple un rol importante en el diagnóstico de la enfermedad de Fabry, ya que aunque la detección de esta por su compromiso renal significaría diagnóstico tardío, debido a que la nefropatía se asocia a complicaciones tardías del fenotipo más severo de la enfermedad.
The early detection of Fabry nephropathy is of interest to us. Its treatment is more effective in early stages. It has been studied by analysing molecular and tissue biomarkers. These have certain disadvantages that hinder its routine use. The aim of this study is to describe the role of the nephrologist in the diagnosis of the disease, and to describe the clinical variables associated with nephropathy in affected patients.
Material and methodsCross-sectional study. Patients were included from three reference centres in Argentina.
ResultsSeventy two patients were studied (26.26±16.48years): 30 of which (41.6%) were men and 42 of which (58.4%) were women; 27 paediatric patients and 45 adults. Fourteen “index cases” were detected, 50% of which were diagnosed by nephrologists. Nephropathy was found in 44 patients (61%): 6 paediatric patients and 38 adults. Two types of clinical variables were associated with nephropathy: (i)peripheral nervous system compromise (P≤.001), angiokeratomas (P≤.001) and auditory compromise (P=.01-.001), with these being early clinical manifestations of the most severe disease phenotype, and (ii)structural heart disease (P=.01-.001) and central nervous system compromise (P=.05-.01), which are major and late complications, responsible for increased morbidity and mortality and lower life expectancy.
ConclusionThe nephrologist plays an important role in the diagnosis of Fabry nephropathy, although the detection thereof owing to its renal involvement would represent a late diagnosis, because nephropathy is associated with late complications of the most severe disease phenotype.
La enfermedad de Fabry (EF, OMIM 301500) es una enfermedad por depósito lisosomal (EDL). Este grupo de enfermedades incluye al menos 50 entidades hereditarias de baja frecuencia, originadas por un error congénito del metabolismo secundario a un defecto monogénico específico que conduce a la deficiencia en la actividad de una enzima lisosomal1. Dicha deficiencia produce el acúmulo de sustratos no metabolizados primariamente en los lisosomas y luego, progresivamente, en otros compartimientos celulares1,2.
En la EF, la actividad ausente o deficiente de la enzimaα-galactosidasa-A (α-gal-A, EC 3.2.1.22) produce la acumulación multisistémica de glicoesfingolípidos complejos, principalmente globotriaosilceramida (Gb3), desde etapas fetales de la vida2.
La incidencia reportada de la EF se encuentra en el rango de un caso por cada 476.000 a un caso por cada 117.000 nacidos vivos en la población general2, aunque cribados neonatales en Italia y Taiwán han reportado resultados mayores3,4. Los estudios de prevalencia en poblaciones susceptibles informan valores de 0,33% en varones y de 0,1% en mujeres con enfermedad renal crónica terminal (ERCT) de etiología desconocida5.
El depósito anormal de sustrato no metabolizado afecta prácticamente a todos los tejidos y órganos de la economía, pero es más predominante en el endotelio y en las células musculares lisas de los vasos sanguíneos, junto con las células epiteliales renales, los cardiomiocitos y las células neurales2. Como consecuencia del almacenamiento lisosomal de Gb3 se desencadena una cascada de fenómenos deletéreos, entre los que se encuentran el compromiso del metabolismo energético, la lesión de pequeños vasos, la disfunción de canales iónicos en células endoteliales, el aumento del estrés oxidativo, las alteraciones de la autofagia, la isquemia y su resultado final, la fibrosis tisular2,6.
La globotriaocilesfingosina (Lyso-Gb3), producto del metabolismo anormal de Gb3, se encuentra elevada en pacientes con EF. Entre sus efectos conocidos se reconocen: a)inhibición de la actividad enzimática α-gal-A; b)liberación local de mediadores químicos de daño glomerular, y c)inducción de proliferación vascular, con engrosamiento del complejo íntima-media arterial7-9.
En el riñón, los depósitos progresivos de Gb3 afectan células tubulares, glomerulares (incluidos los podocitos), endoteliales y musculares lisas vasculares. Esto ha sido demostrado en biopsias renales de pacientes aun sin manifestaciones clínicas de compromiso renal10,11.
Los primeros síntomas de la EF se expresan durante la niñez, con acroparestesias, crisis de dolor neuropático en los cuatro miembros, hipohidrosis, dolor abdominal recurrente, diarrea, náuseas y saciedad precoz, asociados a angioqueratomas. Durante la adolescencia se agregan córnea verticilada, manifestaciones disautonómicas y disminución de la capacidad auditiva. Llegada la adultez, se desarrolla enfermedad renal, cardíaca y cerebrovascular, con morbimortalidad aumentada y disminución de la expectativa de vida comparada con la población general2.
Hasta el año 2001 se describió el fallo renal como la principal causa de muerte en la EF12. Posteriormente se reportó que la principal causa de muerte es la cardiovascular (57% de los casos), que los pacientes que mueren por causa cardiovascular habían recibido previamente terapia dialítica y habían sido diagnosticados en forma tardía13. Por esta razón, la investigación de la nefropatía en los pacientes con EF, sus mecanismos, evolución y tratamiento, ha sido un tema de relevancia entre expertos14-20. Dentro de ese campo de investigación, la detección temprana de la nefropatía ha sido de interés, debido a que los tratamientos específicos para la EF son más eficaces en estadios precoces de daño renal, disminuyendo su eficacia en etapas avanzadas, principalmente debido al desarrollo de fibrosis tisular6,21.
La evaluación de la nefropatía por EF ha sido estudiada por biomarcadores tanto moleculares como tisulares17,22-24, que poseen ventajas y desventajas para su utilización en la práctica habitual; ente las últimas, la escasa accesibilidad de los métodos de alta complejidad en algunas áreas geográficas y las complicaciones de los métodos invasivos dificultan su uso rutinario25. El propósito del presente trabajo es describir el rol del especialista en nefrología en el diagnóstico temprano de la EF y las variables clínicas asociadas a nefropatía en pacientes afectados.
Material y métodosDiseño transversal con recolección de datos retrospectivos. Datos preliminares del presente estudio han sido previamente publicados26.
El estudio fue aprobado por cada comité de ética local. Los pacientes mayores de edad, con criterios de inclusión, firmaron el consentimiento informado. Los menores de edad dieron su asentimiento, y firmó el consentimiento informado su tutor o representante legal de acuerdo con la normativa local.
Se reclutaron pacientes con diagnóstico probable de EF desde junio de 2007 a septiembre de 2017 de tres centros de referencia en Argentina: a)Centro de Neurociencias Los Manantiales, Grupo Gamma Rosario, Rosario, Provincia de Santa Fe; b)Centro de Infusión y Estudio de Enfermedades Lisosomales del Instituto de Nefrología Clínica Pergamino, Pergamino, Provincia de Buenos Aires, y c)Servicio de Terapia Intensiva del Hospital Dr. Enrique Erill de Escobar, Belén de Escobar, Provincia de Buenos Aires. Se incluyeron pacientes con diagnóstico confirmado de EF por estudio genético y dosaje enzimático α-gal-A. Criterios de exclusión: pacientes con nefropatía de otra etiología diferente a la EF. Criterios de eliminación: pacientes con criterio de inclusión que se nieguen a participar del estudio o que presenten alguna complicación relacionada con la extracción de sangre o con la realización de estudios diagnósticos durante su realización. Las mutaciones patogénicas del gen GLA fueron detectadas por estudio genético, mediante secuenciación directa y Multiplex ligation-dependent probe amplification (MLPA)27,28. El dosaje de actividad α-gal-A se realizó por método fluorométrico29, y se consideró normal o disminuido por encima y por debajo de 4,0nmol/h/l, respectivamente. La creatinina en plasma y orina fue determinada por electro-quimio-luminiscencia Roche Diagnostics. El FGe se calculó con ecuación de Schwartz y el CKD-EPI en pacientes menores y mayores de 21años, respectivamente30,31. Para estadificar el FGe se utilizó la clasificación Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline 2013 (KDIGO)32. La albuminuria fue determinada por método colorimétrico Roche Diagnostics33. El cociente albúmina/creatinina en orina fue utilizado para estimar la excreción urinaria de proteínas en 24h. Valores de 0 a 30 fueron considerados normales, de 30 a 300 microalbuminuria y mayor a 300 albuminuria, en al menos dos muestras de orina33. La nefropatía en adultos se definió por: a)microalbuminuria o albuminuria, y/o b)FGe menor a 90ml/min/1,73m2, y/o c)antecedente de requerimiento de terapia de reemplazo de función renal mediante diálisis o trasplante. La nefropatía en pacientes pediátricos se definió por la presencia de excreción urinaria de proteínas aumentada (microalbuminuria o albuminuria); no se consideraron los cambios del FGe por las limitaciones que posee el cálculo del FGe con fórmulas que utilizan creatinina en pacientes pediátricos34. Se determinó la proporción de casos índices diagnosticados por un especialista en nefrología. Se utilizaron las definiciones clásicas de factores de riesgo cardiovascular35. El compromiso del sistema nervioso periférico (SNP) fue considerado por la presencia de dolor neuropático típico de EF o alteraciones de la prueba Quantitative sensory testing (QST)36. El compromiso digestivo fue determinado por la presencia de dolor abdominal, diarrea recurrente, náuseas o saciedad precoz en relación con ingesta copiosa sin otra causa distinta a la EF2,37. La córnea verticilata fue evaluada mediante examen oftalmológico con lámpara de hendidura2,38. Los angioqueratomas fueron evaluados por un dermatólogo especialista en EF2. Las alteraciones auditivas fueron evaluadas por logo-audiometría2. El compromiso cardíaco fue diferenciado en: a)arritmias, y b)cardiopatía estructural, por presentarse ambas alteraciones en la edad pediátrica y adulta, de manera diferencial, en los pacientes afectados. Las arritmias se definieron por la presencia de alteraciones electrofisiológicas en electrocardiograma (ECG) de 12 derivaciones. La cardiopatía estructural se definió por la presencia de hipertrofia del VI en ecocardiograma doppler color y/o imágenes típicas en resonancia nuclear magnética (RMN) cardíaca con gadolinio2,39. El compromiso del sistema nervioso central (SNC) de definió por el antecedente de Stroke y/o imágenes típicas en RMN cerebral asintomáticas2,40.
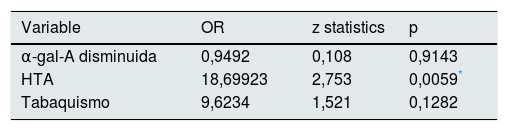
Análisis estadísticoVariable dependiente: nefropatía. Variables independientes: sexo, edad, actividad α-gal-A, compromiso del SNP, compromiso gastrointestinal, córnea verticilata, angioqueratomas, alteraciones auditivas, arritmias, cardiopatía estructural, compromiso del SNC, hipertensión arterial (HTA) y tabaquismo. Se calculó el odds ratio para las dos variables consideradas factores de riesgo de nefropatía en los pacientes con EF: actividad α-gal-A, HTA15,41.
Los datos fueron procesados en una base SPSS Statistics 20.
Para determinar la asociación entre la variable dependiente y las independientes (variables nominales) se utilizó la prueba de chi-cuadrado con datos organizados en tablas de contingencia. No se utilizó factor de corrección de Yates por contar con una muestra mayor a 40, ni test exacto de Fisher por frecuencias esperadas mayor a 5. Se trabajó con intervalo de confianza del 95%. Para el cálculo de p se utilizó la tabla de distribución de chi-cuadrado según grado de libertad. Valores de p<0,05 fueron considerados de significación estadística para rechazar la hipótesis nula (tabla 1).
Distribución de frecuencias y asociación entre nefropatía y las variables independientes estudiadas
| Variable | Valor | Con nefropatía | Sin nefropatía | chi2 | p |
|---|---|---|---|---|---|
| Sexo | Varón | 20 | 10 | 1,03 | 0,3-0,5 |
| Mujer | 23 | 19 | |||
| Edad | Pediátrico | 6 | 21 | 27,49 | < 0,01 |
| Adulto | 38 | 7 | |||
| Actividad α-gal-A | Normal | 21 | 13 | 0,01 | > 0,95 |
| Disminuida | 23 | 15 | |||
| Compromiso del SNP | Sí | 34 | 5 | 13,53 | < 0,001 |
| No | 10 | 18 | |||
| Compromiso digestivo | Sí | 12 | 5 | 0,84 | 0,3-0,5 |
| No | 32 | 23 | |||
| Córnea verticilata | Sí | 12 | 10 | 0,57 | 0,3-0,5 |
| No | 32 | 18 | |||
| Angioqueratomas | Sí | 20 | 6 | 13,53 | < 0,001 |
| No | 24 | 22 | |||
| Compromiso auditivo | Sí | 19 | 3 | 8,5 | 0,01-0,001 |
| No | 25 | 25 | |||
| Arritmias | Sí | 5 | 3 | 0,01 | > 0,095 |
| No | 39 | 25 | |||
| Cardiopatía estructural | Sí | 18 | 2 | 9,72 | 0,01-0,001 |
| No | 26 | 26 | |||
| Compromiso del SNC | Sí | 14 | 2 | 6,03 | 0,05-0,01 |
| No | 30 | 26 | |||
| HTA | Sí | 18 | 1 | 12,28 | < 0,001 |
| No | 26 | 27 | |||
| Tabaquismo | Sí | 6 | 0 | 4,17 | 0,05-0,01 |
| No | 36 | 28 |
HTA: hipertensión arterial; SNC: sistema nervioso central.
Se estudiaron 72 pacientes con diagnóstico confirmado de EF (26,26±16,482años), 30 (41,6%) varones y 42 (58,4%) mujeres. Se incluyeron 27 (37,5%) pacientes pediátricos y 45 (62,5%) adultos.
Fueron encontrados 13 genotipos: E398X, L415P, M296V, L106R, R227Q, A292T, c.448.delG, R363H, C382Y, R301Q, D109G, del 3&4 exons, W81X, todas mutaciones patogénicas del gen GLA. Se estudiaron 14 pacientes «caso índice»: 12 hombres y 2 mujeres, todos adultos. El 50% de los mismos fueron diagnosticados por un especialista en nefrología, el 14,5% por un cardiólogo, el 14,5% por un dermatólogo, y los restantes, el 7% por cada especialidad: neurología, reumatología y genetista. La distribución de frecuencias se muestra en la figura 1. De los 12 casos índices varones, 11 pacientes presentaron daño renal (5 pacientes con ERCT). Las dos mujeres caso índice no presentaron daño renal.

La totalidad de los casos índice presentaron el fenotipo clásico de la enfermedad, habiéndose encontrado las manifestaciones tempranas típicas de la infancia en el 100% de los mismos. La prevalencia de eventos mayores típicos de la edad adulta en la EF en el grupo casos índice fue la siguiente: a)un paciente (6,00%) presentó un evento mayor; b)7 pacientes (46,6%) presentaron dos eventos mayores, y c)6 pacientes (40,0%) presentaron tres eventos mayores.
Se halló nefropatía en 44 pacientes (61,1%): 6 pediátricos y 38 adultos. La media de FGe en la población pediátrica fue 115,81±20,87ml/min/1,73m2, y en adultos, 80,63±42,22ml/min/1,73m2. La figura 2 muestra la distribución de frecuencias de nefropatía según el estadio KDIGO. El 100% de los pacientes en estadios G3, G4 y G5 fueron varones.

El tabaquismo y la HTA fueron los únicos factores de riesgo cardiovascular presentes en la población estudiada.
En la tabla 1 se analiza si existe una relación significativa entre los pacientes con o sin nefropatía (variable dependiente) y una serie de variables clínicas descriptivas (independientes). La tabla 2 muestra los valores de odds ratio calculados para los factores de exposición para la variable dependiente (nefropatía): α-gal-A disminuida, HTA y tabaquismo.
Cálculo de odds ratio para los tres factores de exposición de nefropatía: actividad enzimática, HTA y tabaquismo
| Variable | OR | z statistics | p |
|---|---|---|---|
| α-gal-A disminuida | 0,9492 | 0,108 | 0,9143 |
| HTA | 18,69923 | 2,753 | 0,0059* |
| Tabaquismo | 9,6234 | 1,521 | 0,1282 |
La nefropatía es una de las complicaciones mayores en la EF26. Se caracteriza por proteinuria y disminución progresiva del FGe2,26. La disminución del FGe a través del tiempo se encuentra en relación directa al grado de proteinuria y, sin intervención terapéutica, es más pronunciada en pacientes con FGe inicial menor a 60ml/min/1,73m215. En nuestra población, el 18% de los pacientes presentaron FGe menor a 60ml/min/1,73m2 al momento del diagnóstico. La HTA y el sexo masculino han sido descriptos como factores de riesgo de nefropatía en los pacientes con EF15.
Por la progresión de la nefropatía por EF, los varones afectados presentan ERCT con requerimiento de terapias de reemplazo de función renal a la edad media de 42años14, dato que coincide con la edad de ingreso a diálisis de todos nuestros pacientes con ERCT estudiados.
En la nefropatía por EF se asume que el daño inicial es producido por el depósito anormal de sustrato no metabolizado, Gb3 y sus metabolitos2,20. Esto conduce a una cascada de fenómenos que incluyen el compromiso del metabolismo energético, la lesión de pequeños vasos, la disfunción de canales iónicos en células endoteliales, el aumento del estrés oxidativo, las alteraciones de la autofagia, la isquemia y su resultado final, la fibrosis tisular2,6,20. Lyso-Gb3, el principal metabolito de Gb3, a las concentraciones plasmáticas encontradas en los pacientes con EF induce TGF-β1 autocrina y señales Notch-1 en los podocitos, de manera similar a la respuesta podocitaria a las concentraciones elevadas de glucosa8,42, vías que también conducen a fibrosis renal6.
La terapia de reemplazo enzimático es el único tratamiento específico para la EF que ha demostrado revertir la acumulación tisular de Gb3 en tejido renal, incluidos los podocitos, aunque de manera dosis-dependiente43. Su eficacia es mayor cuanto más tempranamente se inicie, debido a la imposibilidad de corregir la progresión cuando se encuentran presentes lesiones irreversibles, como la fibrosis tisular6,21,44. Chaperonas farmacológicas han demostrado cambios histológicos significativos en células intersticiales capilares, pero no así en podocitos45.
La fibrosis renal ha sido demostrada en pacientes con función renal normal y sin albuminuria10,11,16, y se han estudiado biomarcadores en etapas prealbuminúricas de la nefropatía por EF, tanto moleculares como histológicos8,10,11,16,17,22-24. La alta complejidad y el costo de ciertos métodos no invasivos y las complicaciones de los procedimientos invasivos dificultan su uso rutinario en la práctica habitual, por lo que planteamos un modelo capaz de predecir con validez la asociación entre variables clínicas asociadas a nefropatía y que ayude al médico asistencial en la detección temprana del daño renal en los pacientes afectados, mediante la simple asociación de datos clínicos.
En la evolución natural de la EF el depósito tisular multisistémico de Gb3 se produce de manera progresiva2. Este concepto coincide con los resultados encontrados en nuestro estudio, en el que hubo significación estadística entre nefropatía y la edad de los pacientes. En las primeras etapas del daño tisular los pacientes permanecen asintomáticos, hasta que los depósitos de sustrato y la lesión tisular alcanzan un nivel crítico y se producen manifestaciones clínicas por disfunción orgánica2. Dos tipos de variables clínicas se asociaron con significación estadística a nefropatía en nuestros pacientes: a)compromiso del SNP, angioqueratomas y compromiso auditivo, manifestaciones clínicas del fenotipo clásico (más severo) de la EF, y por otro lado, b)cardiopatía estructural y el compromiso del SNC, que son complicaciones mayores, junto a nefropatía en los pacientes adultos afectados, responsables de la morbimortalidad aumentada y la menor expectativa de vida. Ambos tipos de variables no son manifestaciones tempranas en los pacientes con EF.
ConclusionesLos varones con EF presentan nefropatía avanzada al momento del diagnóstico, y el especialista en nefrología cumple un rol de importancia en el mismo. No hubo diagnósticos de caso índice en etapas pediátricas.
Como se demuestra en este estudio, la asociación de manifestaciones clínicas como variables de relación con nefropatía podría significar el reconocimiento en etapas tardías del daño renal. Parecería razonable continuar en la búsqueda de biomarcadores capaces de diagnosticar nefropatía en etapas precoces, lo cual lograría modificar eficazmente el pronóstico de los pacientes afectados por esta enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés relacionados con el contenido del trabajo enviado.



