




INTRODUCCIÓN
La publicación de una serie de artículos en el número de enero de la revista JAMA bajo el título «Cómo usar un artículo de asociación genética»1-3 constituye un punto de partida excelente para intentar un doble propósito: presentar, a similitud de lo publicado, una guía práctica con los requisitos necesarios para enfrentarse a un artículo de asociación genética y, por otro lado, mostrar las herramientas necesarias para efectuar un estudio de estas características. Para nuestro propósito reduciremos el ámbito de actuación a aquellos trabajos de asociación génica de base poblacional que se realizan a través del reclutamiento de casos y de controles considerando que evaluaremos genes candidatos. Los métodos y la interpretación de los resultados de los estudios genéticos con base familiar son distintos y no serán objetivo de esta revisión. Insistimos en nuestra intención de señalar, únicamente, una serie de pautas de aplicación práctica y no la de pretender la realización de una aproximación a la epidemiología genética. Señalaremos algunas consideraciones sobre la relevancia clínica de estos estudios y analizaremos la situación actual y su supervivencia frente a los estudios de asociación a genoma completo.
¿ESTÁN CORRECTAMENTE SELECCIONADOS LOS PACIENTES? ADECUACIÓN DEL FENOTIPO
La caracterización adecuada del fenotipo asociado con una determinada enfermedad ha de realizarse de acuerdo con aquellos criterios clínicos sobre los cuales haya un consenso médico-científico claramente establecido. La mayor parte de las Sociedades científicas establecen estos criterios y, por supuesto, sus correspondientes actualizaciones, a medida que aumenta el conocimiento sobre el desarrollo y la evolución de la enfermedad. En la realidad de la práctica clínica no siempre es posible establecer un fenotipo certero aunque se sigan rigurosamente estos criterios.
¿ES ADECUADO EL TAMAÑO DE LA MUESTRA?
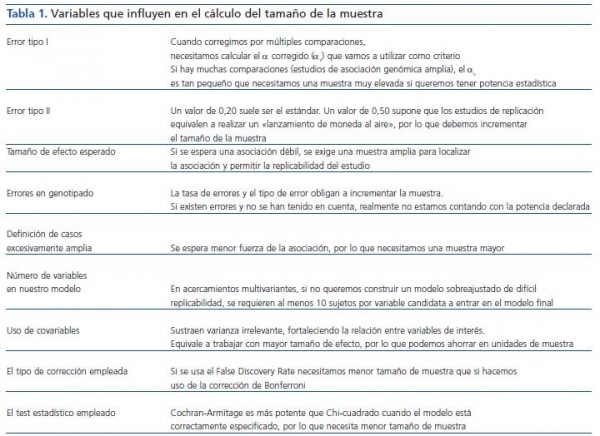
El cálculo del tamaño de la muestra (tabla 1) en un estudio de casos y controles que incluya información genética es objetivo de estudio y modelización constante4. Una aproximación común al cálculo del tamaño de la muestra en un estudio de asociación genética no difiere de la que se realiza en un estudio clínico habitual tipo expuestos/no expuestos y se basa en establecer previamente la magnitud de la diferencia que debe detectarse. En nuestro caso en particular, se traduciría en establecer a priori la diferencia entre la frecuencia alélica o genotípica entre nuestras poblaciones. Asimismo, deberemos conocer la frecuencia de los alelos (todos los que se quieran considerar) en la población control, el valor del error de tipo I, es decir, el error que se comete cuando se rechaza la hipótesis nula siendo verdadera y el valor del error de tipo II, que es el error que se comete cuando se acepta la hipótesis nula siendo falsa. Habitualmente se trabaja con una seguridad de que no cometamos un error del tipo I del 95% (probabilidad, α = 0,05) y del tipo II comprendida entre un 5 y un 20%, si bien, en general, se establece sobre un 20% (probabilidad, β = 0,2). Así nos aseguramos un poder estadístico (1 - β) del 80%. Hay otros aspectos que también debemos considerar, como la tasa previsible de errores y el tipo de errores esperables en nuestro procedimiento de genotipificación, que deben compensarse con un mayor tamaño de la muestra a fin de no ver disminuido nuestro poder estadístico. Algunas herramientas «on-line» nos ayudan realizar estos cálculos. Entre otras: http://linkage.rockefeller.edu/pawe/ y http://hydra.usc.edu/GxE/.
CONSIDERACIONES SOBRE LOS CASOS
De forma ideal se han de reclutar casos incidentes. Conociendo previamente la frecuencia alélica de las variantes que van a estudiarse y antes de proceder a la fase de reclutamiento y conociendo la dificultad inherente a un fenotipo certero, podría intentarse aumentar el número de sujetos «casos» que deben estudiarse a través de distintas alternativas: en primer lugar, incrementando su número siendo menos estrictos en la definición de nuestro fenotipo, por ejemplo, «enfermedad renal », con lo que se aumenta la heterogeneidad y disminuye la certeza de causalidad de los alelos que se evalúan; o bien, por otro lado, podríamos ser más rigurosos en los criterios de selección de fenotipos incrementando la homogeneidad pero reduciendo, necesariamente, el número de casos reclutados. De manera alternativa, puede aumentarse la duración de la fase de reclutamiento. La elección entre una u otra alternativa depende de varios factores, de forma principal de la frecuencia alélica de la variación o de las variaciones en el/los gen(es) candidato que debe estudiarse. Si optamos por utilizar un criterio amplio en la selección de casos, resulta esperable que los genes candidatos estén presentes en un subconjunto inferior de nuestros casos a estudiar; por tanto, debido al menor tamaño de efecto esperado perderemos al menos parte del poder estadístico que esperábamos alcanzar al hacer una definición más amplia de nuestro fenotipo. Por tanto, a la hora de definir los casos debemos ponderar si el incremento de casos disponibles compensa la pérdida del poder estadístico que se deriva de una menor diferencia de frecuencias esperadas. Si la definición del fenotipo la posponemos hasta el momento en que realizamos los análisis de nuestros resultados disponemos de alternativas. Algunos autores, como Chen y Lee, han creado un método cuantitativo y sencillo que nos permite establecer y sistematizar, en función de las frecuencias alélicas y de la existencia de al menos dos posibles tipos de «casos» cuando podemos o no incrementar el tamaño de la muestra de casos uniendo ambos tipos de casos5. No obstante, científicamente lo más adecuado es anticipar estas cuestiones en la fase de diseño del estudio y no cuando procedamos a realizar los análisis estadísticos, ya que podríamos correr el riesgo de sobreajustar los datos y obtener resultados espurios de difícil replicabilidad.
CONSIDERACIONES SOBRE LOS CONTROLES
Arya Sharma y Xavier Jeunemaitre6, reputados autores en el área, señalan una dificultad y un error común en la selección y reclutamiento de la población control. La dificultad deviene de la propia naturaleza de la población de controles. Mientras que para la mayoría de los investigadores hospitalarios la inclusión de pacientes no ha supuesto un problema, sí lo ha sido reclutar controles, ya que requieren de una base poblacional que necesita de recursos específicos. Dado que un control es potencialmente un caso, un sesgo común es incluir a controles de, por ejemplo, bancos de sangre o de trabajadores sanos del medio. La ventaja que teóricamente pudiera suponer la selección de estos candidatos definidos como «hipernormales » ya que, en teoría, redundaría en una mayor diferencia en las frecuencias alélicas entre población afectada y la de controles es, en realidad, mínima y pudiera enmascarar otros fenotipos de selección positiva hacia la supervivencia7-9. De igual forma, la selección de controles que sean casos no diagnosticados supone una reducción del poder estadístico en nuestro estudio10.
SOBRE EL ANÁLISIS ESTADÍSTICO Y LA ASOCIACIÓN ESTADÍSTICA
Podemos establecer una selección del fenotipo que debe estudiarse caracterizándolo como un rasgo dicotómico: diabéticos frente a no diabéticos, hipertensos frente a normotensos o intentar acotar más la variabilidad reduciendo la incertidumbre ambiental y ganando en influencia del genotipo mediante la evaluación de los denominados fenotipos intermedios, es decir, evaluando uno o varios caracteres mensurables que vinculen a través de rutas biológicas plausibles nuestro(s) gen(es) candidato(s) con la enfermedad. La aproximación estadística más convencional a una u otra alternativa implica la realización de modelos multivariantes.
En estudios con múltiples comparaciones debemos utilizar correcciones que nos permitan no incrementar nuestra probabilidad de cometer error tipo I (tabla 2). El procedimiento de Bonferroni consiste en dividir el alfa por el número de comparaciones a estimar: si existen aproximadamente 106 variantes en el genoma, el umbral de valor de p corregida para todas las comparaciones sería p = 0,05/106 = 5 X 10-8. Esto nos obliga a necesitar un tamaño de muestra muy elevado si pretendemos detectar diferencias moderadas. Por ello, se utilizan fórmulas de corrección menos conservadoras, como el False Discovery Rate. No obstante, también con este procedimiento se necesita, en caso de hacer numerosas comparaciones, incrementar de manera prohibitiva el tamaño de nuestra muestra.
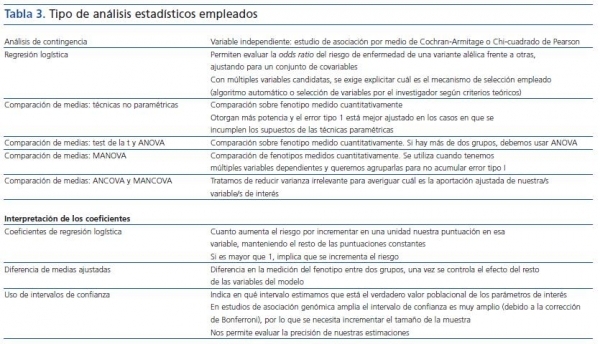
El análisis estadístico utilizado puede otorgarnos mayor poder estadístico (tabla 3). Así, si conocemos el modelo genético (aditivo-recesivo-dominante), debemos utilizar el test de Cochran-Armitage. No obstante, en general no conocemos el modelo genético de nuestros genes candidatos, y aunque nos ofrezca mayor poder, también es cierto que resulta menos robusto que la tradicional prueba de la Chi-cuadrado de Pearson, por lo que en caso de incumplimiento de los supuestos que establezcamos sobre el modelo genético se invalidarían los resultados.
ANÁLISIS DE MEZCLAS POBLACIONALES
Derivado de la teoría evolutiva actual surge la necesidad en medicina de evaluar la heterogeneidad genética de la población en estudio. Los estudios realizados muestran que la variación en la frecuencia de los alelos asociados con la enfermedad entre poblaciones dispares aunque existe es, en realidad, bastante pequeña7. Pudiera ocurrir que la asociación dependiera por completo de la exposición a cierto determinante ambiental cuya frecuencia variase según la localización geográfica y que, por tanto, la frecuencia de estos alelos por selección variase7. Cuando se realizan mezclas poblacionales que difieren en frecuencias alélicas por razones genéticas o ambientales la asociación pudiera resultar espuria. Por tanto, se hace necesaria la selección, genotipificación y análisis de marcadores neutrales (alelos nulos, SNP no ligados, inserciones/deleciones) a través de dos estrategias diferentes denominadas «genomic control» y «structured association»: http://pritch.bsd.uchicago.edu/structure.html y http://wpicr.wpic.pitt.edu/WPICCompGen/genomic_control/genomic_control.htm
ELECCIÓN DE GENES CANDIDATOS
Puesto que en nuestra aproximación hemos decidido evaluar polimorfismos génicos en genes candidatos, nos interesa, en primer lugar, determinar el número y los genes que analizaremos. Tradicionalmente los genes candidatos evaluados se han escogido en función del conocimiento disponible sobre la actividad del producto génico en la enfermedad que debe estudiarse, del conocimiento sobre la función de la proteína codificada por el gen, del derivado de los estudios en modelos animales, del que puede presuponerse a través del fenotipo asociado con las formas monogénicas de la enfermedad, del derivado de estudios de ligamiento génico y de los datos disponibles a priori, así como del obtenido a través de metaanálisis8. Si además las variantes que deben analizarse se sitúan en regiones de interés en los genes, esto se traduce, por ejemplo, en un cambio de aminoácido (variantes no sinónimas), o bien afectan a la estabilidad o al procesamiento del mensajero, o se encuentra en regiones reguladoras del gen, la variante será probablemente, de mayor utilidad11. Una estrategia alternativa en la selección de genes candidatos es la selección de variantes «tags», es decir, variantes de las que se dispone de información previa, obtenida de estudios de ligamiento, y que a su vez pudieran ligarse a alelos de susceptiblidad12.
En un área de trabajo enormemente activa como es la bioinformática, sorprende el escaso número de herramientas que ayudan en la labor clave de la selección de genes candidatos13. Existen, no obstante, una serie de programas on-line: http://omicspace.riken.jp/PosMed/, http://www.genesniffer.org/index/index_frameset.htm y http://www.genetics.med.ed.ac.uk/suspects/. Estos programas integran la información proveniente de estas plataformas de análisis genético de alto rendimiento conjuntamente con la información derivada de los estudios de expresión permitiendo la aparición del término convergencia a la selecciones de regiones y genes candidatos.
FENOTIPO CUANTIFICABLE Y DEMOSTRACIÓN EXPERIMENTAL DEL FENOTIPO CUANTIFICABLE
Identificar y medir el mayor número de parámetros biológicos implicados de manera directa con el gen y su producto o con la ruta biológica en la cual aquel está implicado aumenta sustancialmente la capacidad de información del estudio. Aumenta la rigurosidad si, además, se demuestra de forma experimental que la propia variable se asocia con otras variables en regiones clave del gen o que afecta funcionalmente al gen o a la proteína6.
¿TIENE SENTIDO EL ESTUDIO Y ANÁLISIS DE UN NÚMERO RELATIVAMENTE PEQUEÑO DE POLIMORFISMOS?
Un análisis de un solo polimorfismo puede llevar a asociaciones espurias, entre otros motivos, porque la variante puede encontrarse en desequilibrio de ligamiento con otra u otras variantes, constituyendo un haplotipo característico. Sharma, et al6 consideran que para un gen candidato, la selección, genotipificación y análisis de frecuencia de, al menos, tres polimorfismos comunes permite la identificación de variantes en desequilibrio de ligamiento y la identificación de sinergias. En un estudio de casos y controles de base poblacional en el que se hayan genotipificado SNP próximos y dado que por definición se desconoce la fase, pueden inferirse haplotipos mediante el uso de software genéticos: (GDA: http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php y Arlequín: http://lgb.unige.ch/arlequin/), pero no determinarse en su totalidad ni asociarse con un fenotipo mensurable en un sujeto dado (excepto para los genotipificados como homocigóticos para todos los SNP evaluados en un locus). A día de hoy, la información sobre haplotipos comienza a estar disponible en red y por lo tanto resulta de interés elegir y validar la utilidad pronóstica o terapéutica de variantes funcionales.
Las asociaciones positivas encontradas en una determinada población utilizando una o unas pocas variantes suelen no replicarse en otras poblaciones. La ausencia de replicación constituye un argumento fundamental entre los autores más críticos con los estudios de asociación. Al margen de los problemas derivados de errores de genotipificación, mezclas poblacionales, elección de genes candidatos, caracterización inadecuada de los casos y/o de los controles, reclutamiento inadecuado de los casos y/o de los controles, diferencias en la exposición ambiental, la falta de poder estadístico subyace en la mayor parte de los estudios que muestran ausencia de asociación (tabla 4). La solución no es sencilla, puesto que el reclutamiento de un mayor número de casos y de controles tampoco lo es.
Un estudio de asociación genética retrospectivo de casos y controles debe cumplir por tanto una serie de requisitos sucintamente presentados en los párrafos anteriores.
GENOTIPIFICACIÓN A GRAN ESCALA Y ESTUDIOS DE ASOCIACIÓN GENÓMICA AMPLIA O DE GENOMA COMPLETO
El Proyecto Internacional HapMap (http://www.hapmap.org/index.html.en) se define como un esfuerzo de varios países para identificar y catalogar las similitudes y diferencias genéticas de los seres humanos a este nivel14. Como comentamos al inicio, la metodología y la interpretación de los resultados obtenidos difiere de los análisis de genes candidatos en individuos no relacionados. Las plataformas de análisis genéticos de alto rendimiento ha cambiado el panorama de los estudios de asociación permitiendo genotipificar múltiples polimorfismos aunque necesariamente ampliando el tamaño de la muestra necesaria. La fusión de las tecnologías de genotipificación a gran escala con la información disponible en el proyecto internacional HapMap ha permitido la posibilidad de establecer estudios de asociación génica utilizando la información denominada como de asociación genómica amplia o de genoma completo (WGA). En estos estudios el HapMap provee la información de los denominados «tag SNPs» definidos como el conjunto mínimo de SNP necesarios para detectar un haplotipo. El proyecto ha ido por fases. En la primera se reclutaron tríos (madre, padre e hijos) a partir de los cuales se identificaron SNP localizados a una distancia de 5 kb y con una frecuencia superior al 5%. Caracterizaron entonces su estructura en haplotipos para definir los «tags». En una segunda fase, la identificación de «tags» asociados con una determinada enfermedad permite inferir la estructura del haplotipo disminuyendo la necesidad de genotipificar todas las variantes y permitiendo además localizar genes candidatos próximos. Este tipo de estudios, aunque cada vez más asequibles, requiere a día de hoy de importantes recursos tanto humanos como financieros, pero suponen también un paso importante hacia la caracterización de variables clínicamente relevantes. Sin embargo, la tecnología asociada a la genotipificación no está exenta de problemas15. Incluso cuando se realiza un esfuerzo de esta magnitud son pocas las variantes estadísticamente asociadas a la enfermedad que sobreviven al proceso de replicado16. Surge, además, el problema de las correcciones cuando se realizan múltiples comparaciones estadísticas9,10.
UTILIDAD CLÍNICA
El entusiasmo inicialmente suscitado por los estudios de asociación genética se basó en la facilidad con la que permitían ir un paso más allá sobre la aproximación epidemiológica convencional en el conocimiento de la causalidad de la enfermedad y/o en la de los factores de riesgo asociados a la enfermedad (figura 1). Para uno o varios genes candidatos, la mayoría de los centros hospitalarios podían, por ejemplo, realizar amplificaciones por PCR y digestión enzimática de una serie de polimorfismos en genes de interés una vez reclutados sus casos y controles. Sin embargo, esta información única quedaba sesgada. Esta relativa facilidad permitió un crecimiento exponencial del número de trabajos publicados y, concomitantemente, la aparición de un sector crítico a su utilidad real. Algunos autores señalan además que, puesto que la acción o efecto de una determinada variante dentro de un gen debe interpretarse en el contexto de una red compleja que incluya además de interacciones con otras variantes y con el medio ambiente, la propia complejidad de la ruta biológica en la cual el gen está inmerso, debería plantearse inicialmente la validez de una estrategia de asociación7. En muchos de estos estudios inicialmente realizados, la cuestión crítica fundamental la constituyó la ausencia de reproducibilidad en otras series y poblaciones. Sin embargo, cabe señalar que el denominador común que justifica en muchos casos la falta de reproducibilidad no depende tanto de la población analizada sino, como hemos comentado, de la ausencia de poder estadístico, constituyéndose éste en el error principal. Puesto que la tecnología emergente permite además aumentar de forma exponencial el número de variaciones a analizar es reclutar de manera adecuada la población un requisito fundamental y un problema mayor. En nuestro ámbito, la Ley de investigación biomédica 14/2007, de 3 de julio, regula el tipo de estudios genéticos que pueden realizarse, la estructura de los consentimientos informados necesarios, el proceso de anonimización de las muestras y su almacenamiento, utilización y cesión futura. Por tanto, asociado a los problemas sucintamente presentados en los párrafos anteriores: inadecuada caracterización de la población en estudio, falta de una adecuada evaluación de la población, reclutamiento inadecuado de los casos y/o de los controles, tamaño de muestra insuficiente, falta de replicación de las asociaciones analizadas, se genera un cierto escepticismo que puede, sin embargo, rebatirse con, por ejemplo, una caracterización de fenotipos adecuados, análisis de fenotipos intermedios, evaluación de fenotipos mensurables, caracterizando haplotipos, análisis de la estructura poblacional, de forma que los estudios de asociación genética preserven su cualidad como una de las herramientas de aproximación práctica más poderosas que existen9.

Tabla 1.

Tabla 2.

Tabla 3.

Tabla 4.

Figura 1.



