




Ya han transcurrido veinte años desde la identificación del klotho y del factor de crecimiento fibroblástico 23 (FGF23), el binomio regulador de la homeostasis del fosfato. Al ser el riñón la principal fuente de klotho y el órgano diana regulador del fosfato, la mayoría de los estudios sobre este y el FGF23 tuvieron una vertiente «nefrocéntrica». Gracias al sesgo de este enfoque, los exagerados niveles circulantes de FGF23 observados en la enfermedad renal crónica (ERC) permitieron reconocer el efecto nocivo «off target», independiente de klotho, que ejerce el FGF23. Todo esto ha revolucionado nuestra visión previa sobre la homeostasis mineral y al día de hoy, nos encontramos ante un nuevo escenario en el abordaje clínico del paciente renal, en el que el FGFG23 emerge como marcador precoz de retención de fosfato y simultáneamente como diana terapéutica. En esta revision, se abordan las alteraciones del FGF23 en la ERC y se plantea cómo el mantenimiento del FGF23 circulante en rango adaptativo suprafisiológico desde los estadios iniciales de ERC y el control del «hiperfosfatonismo ilimitado», generado por la resistencia a la acción del FGFG23 en los estadios avanzados, deberían surgir como nuevos paradigmas de tratamiento en chronic kidney disease - mineral and bone disorders (CKD-MBD). El reciente desarrollo de un método automatizado para cuantificar el FGF23, validado para uso clínico, marca el punto de partida para individualizar todo lo que sabemos por los estudios epidemiológicos y utilizarlo adecuadamente desde la cabecera del paciente. Ahora nos toca establecer los límites que discriminen el incremento adaptativo fisiológico de FGF23, para cada estadio de ERC, frente al aumento exagerado, reflejo de una maladaptacion, y que requiera la adopción de medidas terapéuticas.
Twenty years have passed since the identification of klotho and the fibroblast growth factor 23 (FGF23), the regulatory binomial of phosphate homeostasis. Being kidney the main source of klotho as well as a target organ in the phosphate regulation, most studies involving klotho and FGF23 had a «nephrocentric» focus. Considering that circulating FGF23 can reach exaggerated levels at the end stage of chronic kidney disease (CKD), the bias of this approach allowed to recognize the harmful «off target» klotho-independent effect of FGF23. All of these findings have caused a revolution on our previous knowledge about mineral homeostasis and currently, we are facing a new scenario in the clinical management of CKD, where FGF23 emerges simultaneously as an early biomarker of phosphate retention but also as a therapeutic target. In this review, we describe the disturbances of FGF23 in the CKD and we focus on how the maintenance of circulating FGF23 into a supraphysiological adaptive range from the initial stages of CKD and the control of «unlimited hyperphosphatonism» generated by the resistance to FGF23 action at end stages should emerge as new treatment paradigms in chronic kidney disease - mineral and bone disorders (CKD-MBD). The recent development of an automated FGF23 assay, already validated for clinical use, should be the starting point to individualize all our knowledge from epidemiological studies and will allow us to use it properly for the patient's personalized care. Then, now we are in the momentum to assess the discriminating thresholds to distinguish the physiological adaptive FGF23 elevation related to each CKD stage from the exaggerated increase that would be interpreted as a poor regulatory compensation that will requires the adoption of therapeutic intervention.
Han transcurrido veinte años desde la identificación del gen klotho1 y el factor de crecimiento fibroblástico 23 (FGF23) se descubrió poco tiempo después2. La coincidencia entre los fenotipos del ratón con ablación del FGF23 y del ratón klotho condujo al reconocimiento del klotho como correceptor del FGF233 y posteriores trabajos demostraron su vínculo con la homeostasis del fosfato. Esto promovió que, a pesar de haberse descrito el FGF23 en los síndromes hipofosfatémicos familiares, los estudios sucesivos sobre klotho y FGF23 se centraran en la enfermedad renal crónica (ERC), al ser el riñón la principal fuente de klotho y, al mismo tiempo, órgano diana en la regulación del fosfato. Todos ellos han revolucionado nuestro enfoque previo sobre la homeostasis mineral y nos situan ante un nuevo escenario en el abordaje clínico del paciente renal, en el que el FGF23 emerge como marcador precoz de retención de fosfato y, al mismo tiempo, como diana terapéutica.
Tomando conciencia de esta revolución, en el año 2006, el grupo de trabajo de kidney disease: improving global outcomes (KDIGO) introduce la nomenclatura de chronic kidney disease - mineral and bone disorders (CKD-MBD)4 para designar las alteraciones de la homeostasis mineral que caracterizan a la ERC y limita la utilización del término «osteodistrofia renal» a las anomalías óseas desencadenadas por este desequilibrio. La finalidad de este cambio es resaltar la precocidad de estas alteraciones que, al iniciarse desde el «minuto uno» de ERC, excluyen el paradigma previo de complicación tardía. Simultáneamente, se amplía su ámbito al incluir la participación de otros tejidos extra-óseos con la aparición de calcificación de partes blandas, especialmente a nivel vascular. Precisamente, el fósforo adquiere protagonismo por su papel clave en el desarrollo de este envejecimiento y calcificación vascular5 y se empieza a asumir que un elemento esencial y tóxico, en un margen tan estrecho, no puede estar supeditado al calcio ni controlado por las hormonas calciotrópicas. En este escenario, el binomio FGF23/klotho resulta ser la pieza que faltaba en ese complejo rompecabezas que representa la regulación endocrina de la homeostasis mineral.
El FGF23 es una proteína ósea que forma parte de la subfamilia de los factores fibroblásticos (FGF) endocrinos. Aunque comparte la estructura «core» común a todos los FGF, el FGF23 se diferencia por un segmento característico de escisión entre la secuencia de aminoácidos 176 a 179, delimitado por dos residuos de arginina (Arg). Tras su transcripción, regulada principalmente por la sobrecarga crónica de fosfato, el calcitriol y la parathormona (PTH), el FGF23 sufre otros dos procesos intra-óseos a nivel de ese segmento: una glicosilación mediada por la enzima N-acetil-galactosaminil-transferasa3 (GALNT3)6 que lo estabiliza y que limita un segundo proceso de degradación regulado por proteasas, sobretodo por furinas. De este modo, los niveles circulantes de FGF23 representan el balance entre su transcripción y su degradación, detectándose en la sangre la molécula intacta de FGF23 biológicamente activa junto con una serie de fragmentos de FGF23 inactivos, fruto de esa escisión. A nivel fisiológico, sus órganos diana son aquellos que expresan klotho: riñón, paratiroides y encéfalo; una proteína que forma complejos binarios con el receptor FGF1 (FGFR1), incrementando su afinidad y especificidad por el FGF23. De este modo, al señalizar ras-mitogen-activated protein kinase (MAPK/RAS), el binomio FGF23/klotho regula el fosfato, promoviendo su excreción urinaria al reducir la expresión de los co-transportadores NaPi2a y 2c en la membrana del túbulo proximal y su reabsorción. Al mismo tiempo, impide una nueva sobrecarga de fósforo procedente del intestino o por un incremento en la resorción ósea, mediante la acción supresora de la vitamina D y la PTH7 (fig. 1 A). Como consecuencia de este efecto supresor, disminuyen indirectamente los niveles de calcio y por ello, ante situaciones de hipocalcemia con riesgo de compromiso vital, el organismo se defiende, inhibiendo la síntesis de FGF238. A nivel del encéfalo se desconoce su acción, aunque se ha relacionado con las funciones cognitivas.
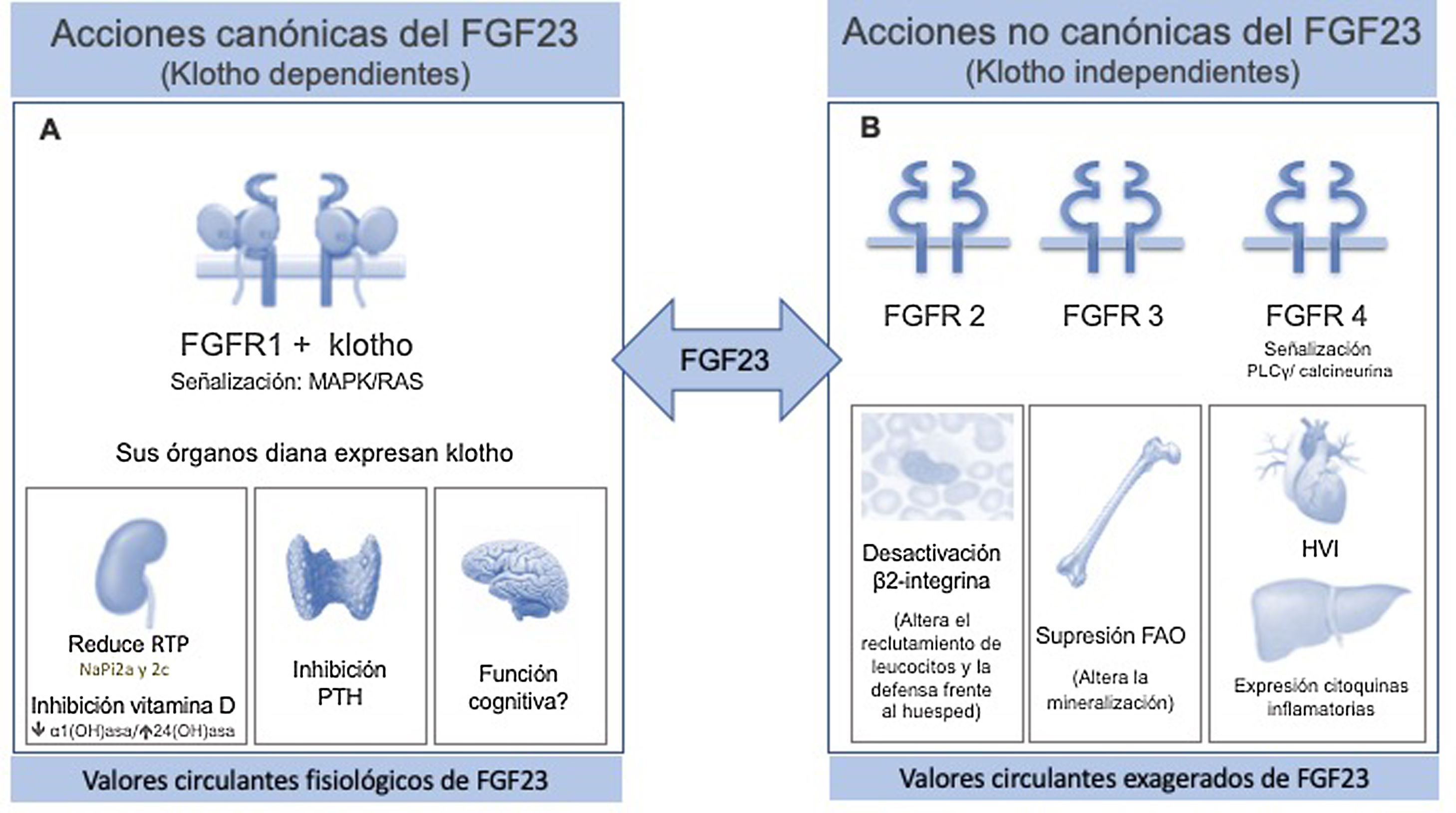
A) Acción fisiológica klotho dependiente. El FGF23 controla la homeostasis del fósforo, señalizando el receptor 1 (FGFR1) con la participación de la proteína klotho que actúa como correceptor, incrementando la sensibilidad y la especificidad del FGFR1 para el FGF23; por ello, sus órganos diana son aquellos que expresan klotho. En riñón ejerce una acción fosfatúrica, al inhibir los cotransportadores sodio-fosfato 2a y 2c, liberando el exceso de fosfato; simultáneamente inhibe la síntesis de calcitriol al suprimir la 1?-hidroxilasa y promueve su catabolismo por la 24-hidroxilasa. Este efecto sobre la vitamina D, unido al efecto supresor sobre la PTH en la paratiroides, impide nueva entrada de fosfato al organismo procedente de intestino y hueso. A nivel encefálico se desconoce su acción, aunque se relaciona con las funciones cognitivas.
B) Acción tóxica independiente de klotho. Los niveles exageradamente elevados de FGF23 que se evidencian en los últimos estadios de ERC, son capaces de activar otros receptores en otros órganos que no expresan klotho, ocasionando importantes patologías. Como ejemplo, mediante la señalización del FGFR2 desactiva la β2-integrina, alterando el reclutamiento de los leucocitos y la defensa frente al huésped, o suprime a nivel óseo la expresión de la fosfatasa alcalina ósea a través de la activación del FGFR3, alterando la mineralización por el acúmulo de pirofosfato. A través de la señalización del FGFR4 el FGF23 ejerce un efecto tóxico directo sobre el miocardio, induciendo hipertrofia ventricular izquierda y sobre el hígado induce la expresión de citoquinas inflamatorias, empeorando aun más el estado inflamatorio que caracteriza al enfermo renal e incrementando la síntesis de FGF23.
ERC: enfermedad renal crónica; FGF23: factor de crecimiento fibroblástico 23; FGFR1: receptor uno del factor de crecimiento de fibroblastos; FGFR2: receptor dos del factor de crecimiento de fibroblastos; FGFR3: receptor tres del factor de crecimiento de fibroblastos; FGFR4: receptor cuatro del factor de crecimiento de fibroblastos; PTH: parathormona.
El desarrollo de métodos para cuantificar el FGF23 circulante permitió establecer su implicación en la ERC. Los primeros enzimoinmunoensayos (EIA), no validados para uso clínico y aun vigentes, se clasificaron en dos tipos de formato, dependiendo de su configuración antigénica (tabla 1). El formato conocido como FGF23 intacto (iFGF23), que reconoce exclusivamente la molécula completa y activa de FGF23, refleja mejor su actividad biológica, aunque no es equivalente, ya que los fragmentos carboxi-terminales también se unen al correceptor klotho y, al competir con el iFGF23 reducen su señalización9. Su principal inconveniente es el efecto de una posible degradación ex vivo por las proteasas del suero, siendo por ello, el plasma la matriz biológica de elección. Al mismo tiempo, muestra una mayor variabilidad intraindividual por la influencia de su ritmo circadiano ya que, como cualquier proteína ósea, el FGF23 sufre un incremento nocturno que culmina con un pico hacia las ocho de la mañana10. Como contrapartida, el formato de EIA carboxi-terminal (cFGF23), que reconoce tanto la forma activa como los fragmentos C-terminales, es reflejo de la transcripción ósea del FGF23 y presenta una mayor estabilidad analítica. Sin embargo, su variabilidad interindividual es mayor, dificultando el establecimiento de intervalos biológicos de referencia. Ambos métodos presentan ventajas e inconvenientes, incluso podría ser interesante cuantificarlos conjuntamente para identificar la relación entre el FGF23 activo y sus fragmentos. En cualquier caso, gran parte de lo que hoy sabemos sobre la CKD-MBD se debe a la utilización de estos EIA, cuyo potencial uso en el manejo clínico de la ERC ofrece importantes ventajas sobre los marcadores bioquímicos clásicos de metabolismo mineral.
Aspectos pre-analíticos y analíticos en la medición de FGF23
| Aspectos pre-analíticos de la cuantificación de FGF23 |
|---|
| Ritmo circadiano con pico de secreción a las 8 a. m. Extracción idónea: 8-10 a. m. |
| Posible degradación in vitro por proteasas. Matriz biológica: plasma |
| Aspectos analíticos de la cuantificación de FGF23 |
| Método FGF23 intacto (iFGF23) | Método FGF23 C-Terminal (C-FGF23) |
|---|---|
| Mide exclusivamente forma intacta del FGF23 | Forma intacta + fragmentos carboxiterminales |
| Refleja transcripción + proteolisis intraósea | Refleja exclusivamente transcripción ósea |
| Representa la actividad biológica del FGF23 | Representa concentración, pero no actividad |
| Marcado ritmo circadiano | Poco influido por el ritmo circadiano |
| Mayor variabilidad intra-individual | Mayor variabilidad inter-individual |
| Menor estabilidad analítica (degradación in vitro) | Menos afectado por la degradación in vitro |
FGF23: factor de crecimiento fibroblástico 23.
La primera ventaja es la precocidad de su elevación. En el estadio uno de ERC, ya comienza a evidenciarse la disminución en la expresión renal de klotho11, cuyo gen es muy sensible a la inflamación y a la agresión renal12. Conforme progresa la ERC, junto con la pérdida de nefronas funcionales, se produce una inhibición en la expresión de klotho por la contribución de otros factores, entre ellos, las propias toxinas urémicas que silencian su gen por hipermetilación13, el incremento en la excreción fraccionada de fósforo14, la angiotensina II15 o la deficiencia de vitamina D16. De este modo, al ser el riñón la principal fuente de klotho del organismo, la ERC representa el prototipo de la deficiencia adquirida de klotho. El problema es que, al mismo tiempo, es órgano diana y, ante un riñón resistente por la pérdida de nefronas y de klotho, el organismo responde incrementando la síntesis ósea de FGF23 para mantener la normofosfatemia. Esta respuesta se inicia en las fases precoces de ERC, con filtrados glomerulares (FG) de alrededor de 75 mL/min y antecede al resto de biomarcadores (fig. 2). De hecho, los valores más elevados de FGF23 son predictivos de hiperparatiroidismo secundario (HPTs)17, situación lógica, ya que este resulta de la inhibición de la vitamina D promovida por el propio FGF23 y de su correspondiente hipocalcemia. Junto con este incremento compensador, existen otros factores que adicionalmente pueden modular la expresión ósea de FGF23 en la ERC. La suplementación con hierro parenteral induce su síntesis; efecto que se vincula con algunos excipientes como la sacarosa o la polimaltosa18, ya que no se produce con todos los preparados parenterales ni con el formato oral. Por el contrario, la deficiencia de hierro, la anemia y en general las situaciones de hipoxia, también promueven la transcripción ósea de FGF23 a través de la inducción y la estabilización del factor inducido por hipoxia 1α (HIF1α)19. Finalmente, y de manera muy especial, la propia tormenta inflamatoria que caracteriza a la ERC va a ejercer un importante impacto sobre la expresión ósea de FGF23 al condicionar una deficiencia funcional de hierro. Ante esta situación, se incrementa la expresión y estabilización del HIF1α y, como consecuencia, la transcripción ósea de FGF23; sin embargo, el HIF1α aumenta simultáneamente la expresión de furinas y la proteólisis del FGF23 para evitar una señalización FGFR1 innecesaria debida al incremento de FGF23 activo. Lógicamente, este aumento en la transcripción de FGF23 promovido por la inflamación solo podríamos detectarlo con la utilización de un método cFGF23 ya que, al degradarse en fragmentos, no es evidenciable al emplear un método iFGF23. Sin embargo, cuando se cronifica la inflamación, se agota esta proteólisis compensadora y se elevan los niveles circulantes de iFGF23, un mecanismo proteolítico compensador que también esta alterado en el paciente renal20 y por esto, en estas situaciones, los dos métodos para cuantificar FGF23 tienen la misma validez.
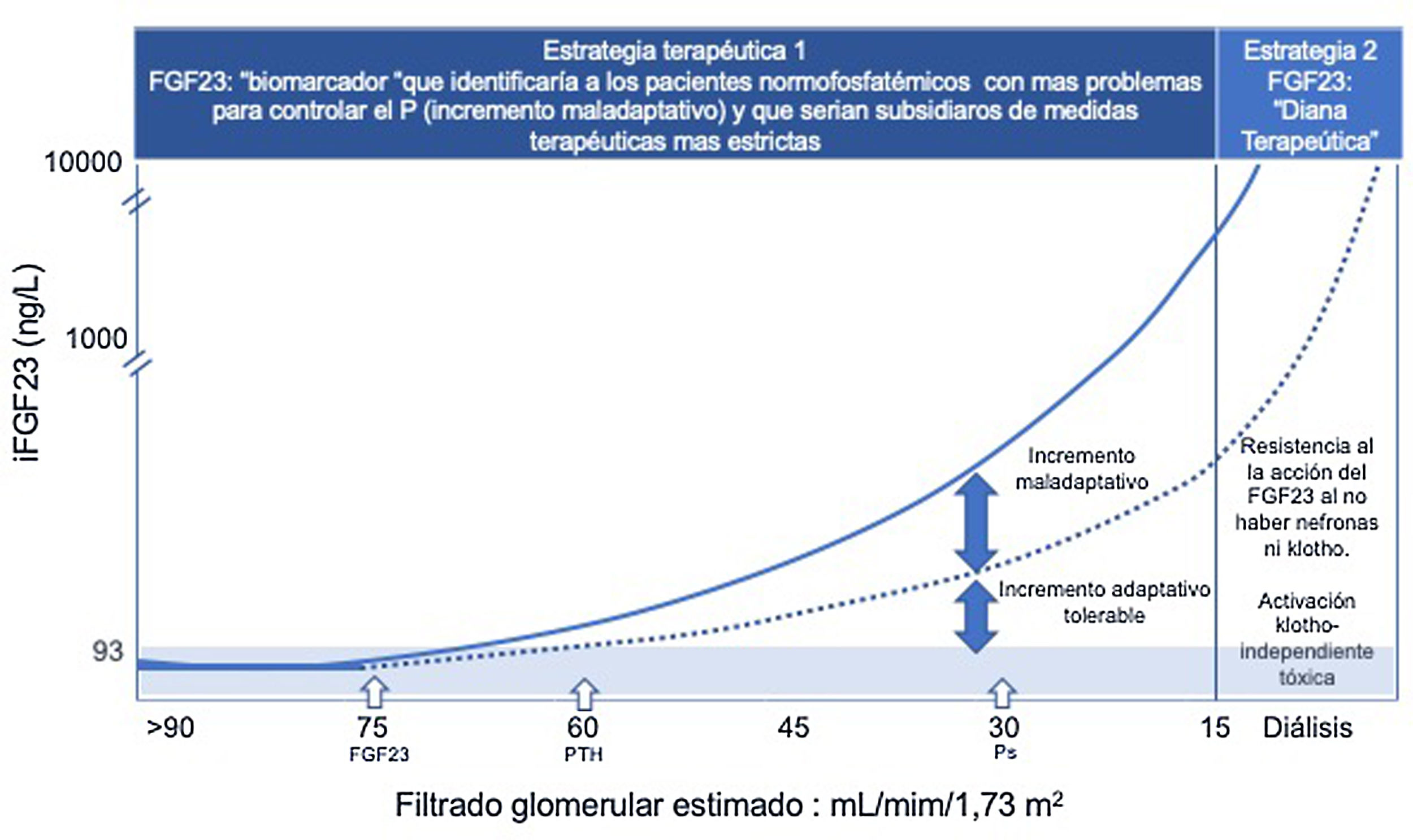
FGF23 biomarcador precoz de retención de fosfato y diana terapéutica.
Estrategia terapéutica 1: Ante la pérdida de nefronas y de klotho, se produce precozmente un incremento adaptativo de FGF23 (eFG: 75 mL/min/ 1,73 m2) para mantener la normofosfatemia. Cuando se sobrepasa la capacidad fosfatúrica compensadora del FGF23 y de la PTH aparecen las situaciones de hiperfosfatemia (eFG: 30 mL/min/ 1,73 m2), pero es tarde porque el efecto tóxico vascular del fosfato ya se ha producido.
La precocidad de su elevación, unida a su valor predictivo de mortalidad en condiciones de normofosfatemia, avalan la utilidad de la cuantificación de FGF23, especialmente en los primeros estadios de la ERC, permitiendo identificar a aquellos pacientes normofosfatemicos con un «incremento maladaptativo», reflejo de mayores problemas para controlar el fósforo, y que requerirían la adopción de medidas terapéuticas más estrictas.
Estrategia terapéutica 2: En los estadios finales de ERC, la ausencia de nefronas y de klotho generan una resistencia a la acción del FGF23 y los niveles circulantes de FGF23 son ilimitados. Estos valores exagerados de FGF23 son capaces de activar otros receptores FGFR produciendo patología en otros órganos y sistemas que no expresan klotho. En esta situación el FGF23 se convierte en diana terapéutica para evitar su toxicidad.
ERC: enfermedad renal crónica; FGF23: factor de crecimiento fibroblástico 23; FGFR: receptor del factor de crecimiento de fibroblastos; PTH: parathormona.
Como sucede con otras moléculas, el incremento de los niveles circulantes de FGF23 se asocia con una progresión más rápida de la ERC21. Los hiperfosfatonismos más acentuados, generados como respuesta compensadora para mantener la normofosfatemia, reflejan mayores problemas para controlar el fósforo en estos primeros estadios. A su vez, el mayor esfuerzo fosfatúrico que induce el FGF23 pasa factura a la propia nefrona, como se menciona posteriormente22 y contribuye a una mayor pérdida en la expresión de klotho14. Similarmente, los niveles exageradamente aumentados de FGF23 en la ERC se vinculan con la mortalidad23. La ventaja de esta última relación sobre el resto de los biomarcadores es que, además de ser creciente conforme se aplica el análisis multivariante, es significativa en situaciones de normofosfatemia. Y esto es sumamente importante porque el fosfato es un marcador insensible, que solamente se eleva cuando se sobrepasa la capacidad compensadora de las hormonas fosfatúricas (FGF23 y PTH) (fig. 2), situación en la que cualquier actitud terapéutica llega tarde porque ya se ha producido ese proceso de envejecimiento y de calcificación vascular. En este sentido, la monitorización del FGF23 surge como una valiosa herramienta diagnóstica y pronóstica al permitir identificar a aquellos pacientes que, aun mostrando valores de fosfatemia en rango biológico de referencia, comienzan a tener más problemas para controlar la homeóstasis del fósforo.
La aportación del FGF23 como indicador de retención precoz de fosfato implica una nueva orientación en el tratamiento del paciente al ofrecernos la posibilidad de adelantarnos y controlar oportunamente la CKD-MBD, aplicando estrategias terapéuticas reservadas a fases más avanzadas de ERC, como los captores no cálcicos. El propio FGF23 se convierte en diana terapéutica y el objetivo actual del tratamiento ya no sería corregir la hiperfosfatemia que aparece en los estadios avanzados, sino garantizar un adecuado control del fosfato desde los estadios iniciales de ERC, evitando el incremento de los valores circulantes de FGF23 más allá de un rango adaptativo suprafisiológico. Los niveles elevados de FGF23 implican un mayor esfuerzo fosfatúrico en las nefronas funcionantes, concentrándose gran cantidad de fosfato en la luz tubular. Este precipita con el calcio y se une a la fetuina-A, formando las denominadas partículas de calciproteina, nanopartículas que, a su vez, producen citotoxicidad en la célula tubular y más daño renal22. Estudios muy recientes señalan un incremento de estas partículas de calciproteina dispersas en la sangre de los pacientes renales que inducirían más respuesta inflamatoria y calcificación vascular y que, al mismo tiempo, tras depositarse en el hueso activarían la expresión ósea de FGF2324. Por todo ello, la aplicación de medidas dietéticas más restrictivas, con una ratio fósforo/proteína favorable para no desnutrir, la utilización de captores no cálcicos con ventajas frente a los cálcicos en la reducción del hiperfosfatonismo25 y el control de otros estímulos no adaptativos como la anemia o la administración IV de preparados de hierro con sacarosa o polimaltosa, en el momento en el que se sobrepase ese incremento adaptativo, teóricamente minimizaría el impacto del fosfato sobre la morbi/mortalidad del paciente renal. El beneficio de un correcto control de la CKD-MDB desde el inicio de la ERC va a condicionar la evolución de un futuro transplante, ya que los niveles más elevados de FGF23 y PTH previos a este son predictivos de hiperfosfatonismo e hiperparatiroidismo terciario persistentes; situaciones que predisponen a una mayor pérdida de masa ósea y a un incremento en el riesgo de pérdida del injerto y/o de mortalidad26,27.
Potenciales beneficios de la cuantificación de FGF23 en la ERC avanzadaEn las fases avanzadas de ERC, cuando apenas quedan nefronas y klotho, se descompensa este mecanismo adaptativo y se produce una resistencia a la acción del FGF23; es más, la presencia del HPTs supone un estímulo añadido a esta situación (fig. 2). Los valores circulantes de FGF23 pueden llegar a ser más de 1.000 veces su rango de referencia biológico, pasando factura al organismo. Ante esa pérdida de la señalización canónica klotho-dependiente, los valores casi ilimitados de FGF23 activan otros receptores diferentes del FGFR1, ocasionando patología en órganos que no expresan klotho (fig. 1B); una activación que, posiblemente, se ve facilitada por la presencia de otras proteínas que actuarían como correceptores. De este modo, ante la ausencia del efecto protector del klotho, el FGF23 activa en el miocardio el receptor cuatro del factor de crecimiento de fibroblastos (FGFR4) y, al señalizar fosfolipasa C/calcineurina, incrementa los niveles citoplasmáticos de calcio e induce hipertrofia ventricular izquierda (HVI)28,29. Curiosamente, los pacientes con raquitismo hipofosfatémico familiar ligado al cromosoma X (XLH) no presentan HVI a pesar del aumento de FGF23; el motivo de esta discrepancia se debe probablemente a la contribución de otros factores, que no están presentes en el fenotipo XLH, como la hiperfosfatemia y la deficiencia de klotho, que favorece la unión del FGF23 al FGFR4 y eleva la vulnerabilidad del miocardio al perderse los efectos protectores del klotho soluble sobre el sistema cardiovascular30. Este efecto tóxico directo sobre el miocardio se ve agravado por otros efectos indirectos del FGF23 como son el incremento en la expresión y en la actividad renal del co-transportador sodio/cloro (NCC), que al retener sodio aumenta el volumen plasmático y el daño hipertensivo31, o la supresión en la expresión de la enzima convertidora dos de angiotensina (ACE2) y de su efecto beneficioso contrarregulador del sistema renina angiotensina (RAS)32. A través de la activación del receptor tres del factor de crecimiento de fibroblastos (FGFR3), el FGF23 ejerce una acción supresora autocrina sobre la transcripción ósea de fosfatasa alcalina no específica de tejido (TNAP) y, al acumularse el pirofosfato, se genera un defecto en la mineralización33. Los niveles excesivos de FGF23 también activan el receptor dos del factor de crecimiento de fibroblastos (FGFR2) en los neutrófilos, afectándose la β2-integrina, el reclutamiento de los leucocitos y la defensa frente al huésped; situación que justifica la mayor susceptibilidad a infecciones que muestra el paciente renal34. Al margen de otros efectos, el FGF23 también tiene impacto sobre el hepatocito, ya que expresa FGFR4 y su activación extra-canónica por el FGF23 promueve la expresión de citoquinas inflamatorias (IL6) y de proteína C reactiva (PCR), autoperpetuando la inflamación y cerrando un círculo vicioso35. Es más, como dato curioso, se ha demostrado producción ectópica de FGF23 en las células hepáticas de pacientes con poliquistosis renal autosómica dominante, lo que explica los valores significativamente más elevados de FGF23 plasmático que muestran estos casos en relación con otros enfermos renales con una tasa semejante de FG36.
En estas fases avanzadas de ERC, el FGF23 sigue siendo diana terapéutica y el objetivo sería minimizar el efecto de su toxicidad sobre otros órganos y sistemas. A diferencia de los estadios iniciales donde prima el mantenimiento de los niveles circulantes de FGF23 dentro de su rango adaptativo, reflejo de un adecuado control del fósforo y de la CKD-MBD, en los pacientes renales en estadio cinco, la estrategia terapéutica se basaría en reducir su señalización, dado que el control del hiperfosfatonismo es sumamente complicado. Esto resulta sorprendente a priori, ya que estudios previos de experimentación animal demuestran que la neutralización del FGF23 en la ERC tiene beneficios sobre el HPTs, pero a costa de un incremento en la mortalidad37; una situación que se comprende fácilmente porque al neutralizar al FGF23 se bloquea su importante mecanismo compensador sobre la CKD-MBD y el resultado es un aumento incontrolado del fosfato con el consiguiente impacto tóxico sobre el envejecimiento vascular y la mortalidad. Por esto, esta estrategia terapéutica está totalmente contraindicada mientras quede riñón funcionante sobre el que pueda actuar el FGF23; sin embargo, en estas fases finales en las que los mecanismos fosfatúricos adaptativos ya no tienen sentido al no haber ni klotho ni nefronas, sí sería planteable la reducción de esa señalización no canónica del FGF23 que solo ejerce efectos nocivos sobre el organismo. Esta hipótesis de tratamiento abre un nuevo horizonte por explorar. Recientemente, se ha desarrollado un anticuerpo IgG1 monoclonal humano: burosumab, dirigido contra la molécula de FGF23, indicado en los síndromes XLH con muy buenos y prometedores resultados38. Otra estrategia podría ser el bloqueo selectivo de los receptores específicamente involucrados en los efectos adversos inducidos por FGF23. En este sentido, la utilización de anticuerpos anti-FGFR4 podría ser otra buena aproximación para reducir la inflamación, la HVI y la morbi/mortalidad en la ERC35.
De la nefrología de salón a la cabecera del pacienteTodo lo anteriormente expuesto avala la potencial utilidad de la cuantificación de FGF23 en la ERC. Sin embargo, los primeros EIA no llegaron a implementarse en los laboratorios clínicos por tratarse de métodos no automatizados y no validados para uso clínico. En la actualidad, ya disponemos de uno de ellos. Se trata de un método quimioluminescente (ECLIA) que cuantifica iFGF23. Existen diversos estudios que validan su comportamiento analítico y lo evalúan tanto en la población general como en pacientes con ERC39,40. Tras varios años demandando un método FGF23 para uso clínico, este ECLIA iFGF23 automatizado marca el punto de partida para individualizar todo lo que sabemos por los estudios epidemiológicos y, sin embargo, curiosamente, no termina de implementarse en la práctica diaria. Su precio elevado y la ausencia de guías clínicas que recomienden su cuantificación podría ser parte de este problema, pero hay que tener en cuenta dos argumentos. En primer lugar, que, por definición, cualquier método que no esté automatizado no se contempla en ninguna guía y, por tanto, los EIA anteriormente desarrollados no tuvieron opción. En segundo lugar, que la falta de experiencia y de estudios con este nuevo método impide establecer rangos y recomendaciones sobre su utilización en el contexto de la CKD-MBD. Resumiendo, la situación actual es que tenemos la pelota en nuestro tejado y es el momento de trabajar con este prometedor biomarcador para responder a muchas de las preguntas que aún nos hacemos y, una vez respondidas, comencemos a emplearlo adecuadamente en el manejo de la CKD-MBD.
En población renal, los estudios de validación previamente mencionados39,40 demuestran esa esperable asociación inversa entre el incremento del iFGF23 y el FG, y establecen unos valores medios y rangos intercuartiles para cada estadio de ERC. Lógicamente, estos rangos son superiores a los observados en la población general ya que la elevación del FGF23 supone un mecanismo de adaptación de la CKD-MBD. Podemos considerar a estos rangos como una aproximación; el problema es que, incluyen tanto a los pacientes con una buena adaptación como a los maladaptados. De este modo, el primer objetivo que planteamos sería la identificación de ese valor de decisión clínica para cada estadio de ERC, es decir, el umbral específico que delimita el incremento adaptativo fisiológico esperable del aumento exagerado patológico, con la finalidad de poder discriminar a aquellos pacientes mal controlados. Respondiendo, en parte, a esta cuestión clave aun no contestada, hay un estudio recientemente publicado41 en una gran cohorte de pacientes con ERC avanzada, que establece un posible punto de corte para este ECLIA iFGF23 automatizado, dentro de ese rango elevado de FGF23 para cada estrato de FG, a partir del cual se incrementa el riesgo de eventos cardiovasculares y renales. Sus resultados indican que el umbral óptimo de riesgo para el iFGF23 se eleva conforme declina el FG y que el umbral de riesgo cardiovascular (177 ng/L para eFG ≥ 30 mL/min; 228 ng/L para eFG 20 a 29 mL/min y 528 ng/L para eFG < 20 mL/min) es diferente al del riesgo renal (141 ng/L para eFG ≥ 30 mL/min; 192 ng/L para eFG 20 a 29 mL/min y 311 ng/L para eFG < 20 mL/min). Este trabajo representa el primer intento para abordar este reto, ofreciendo nuevos criterios para el manejo clínico del paciente con ERC avanzada y para futuros ensayos clínicos. Pero, fundamentalmente, invita a realizar nuevos estudios que, además de avalar sus hallazgos, contemplen otras poblaciones y otros aspectos no evaluados.
Considerando que los umbrales óptimos de riesgo difieren según el tipo de evento sería interesante evaluar la asociación de este ECLIA con otro tipo como el desarrollo de HPTs o, muy especialmente, con la calcificación vascular. Asimismo, aunque estos puntos de corte con valor pronóstico de riesgo se establezcan para cada estadio de ERC, el carácter precoz de la elevación del FGF23 hace muy interesante su evaluación en los estadios 3a y 3b de ERC con la idea de anticipar las posibles estrategias terapéuticas a los pacientes en riesgo. Una vez establecidos estos umbrales específicos de riesgo, habrá que demostrar si la corrección de estos valores aberrantemente elevados de iFGF23 aporta beneficios en la morbi/mortalidad del enfermo renal. Aceptemos este reto para avanzar de la nefrología de salón a la cabecera del paciente.
FinanciaciónProyecto financiado por el Fondo de investigación sanitaria ISC-III y cofinanciado con fondos FEDER PI16/01298.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



