



Twenty years have passed since the identification of klotho and the fibroblast growth factor 23 (FGF23), the regulatory binomial of phosphate homeostasis. Being kidney the main source of klotho as well as a target organ in the phosphate regulation, most studies involving klotho and FGF23 had a “nephrocentric” focus. Considering that circulating FGF23 can reach exaggerated levels at the end stage of chronic kidney disease (CKD), the bias of this approach allowed to recognize the harmful “off target” klotho-independent effect of FGF23. All of these findings have caused a revolution on our previous knowledge about mineral homeostasis and currently, we are facing a new scenario in the clinical management of CKD, where FGF23 emerges simultaneously as an early biomarker of phosphate retention but also as a therapeutic target. In this review, we describe the disturbances of FGF23 in the CKD and we focus on how the maintenance of circulating FGF23 into a supraphysiological adaptive range from the initial stages of CKD and the control of “unlimited hyperphosphatonism” generated by the resistance to FGF23 action at end stages should emerge as new treatment paradigms in CKD-MBD. The recent development of an automated FGF23 assay, already validated for clinical use, should be the starting point to individualize all our knowledge from epidemiological studies and will allow us to use it properly for the patient’s personalized care. Then, now we are in the momentum to assess the discriminating thresholds to distinguish the physiological adaptive FGF23 elevation related to each CKD stage from the exaggerated increase that would be interpreted as a poor regulatory compensation that will requires the adoption of therapeutic intervention.
Ya han transcurrido veinte años desde la identificación del klotho y del Factor de crecimiento fibroblástico 23 (FGF23), el binomio regulador de la homeostasis del fosfato. Al ser el riñón la principal fuente de klotho y el órgano diana regulador del fosfato, la mayoría de estudios sobre klotho y FGF23 tuvieron una vertiente “nefrocéntrica”. Gracias al sesgo de este enfoque, los exagerados niveles circulantes de FGF23 observados en la enfermedad renal crónica (ERC) permitieron reconocer el efecto nocivo “off target” independiente de klotho que ejerce el FGF23. Todo esto ha revolucionado nuestra visión previa sobre la homeostasis mineral y a día de hoy, nos encontramos ante un nuevo escenario en el abordaje clínico del paciente renal, en el que el FGFG23 emerge como marcador precoz de retención de fosfato y simultáneamente como diana terapéutica. En esta revisión se abordan las alteraciones del FGF23 en la ERC y se plantea cómo el mantenimiento del FGF23 circulante en rango adaptativo suprafisiológico desde los estadios iniciales de ERC y el control del “hiperfosfatonismo ilimitado”, generado por la resistencia a la acción del FGFG23 en los estadios avanzados, deberían emerger como nuevos paradigmas de tratamiento en la CKD-MBD. El reciente desarrollo de un método automatizado para cuantificar FGF23, validado para uso clínico, marca el punto de partida para individualizar todo lo que sabemos por los estudios epidemiológicos y utilizarlo adecuadamente desde la cabecera del paciente. Ahora nos toca establecer los límites que discriminen el incremento adaptativo fisiológico de FGF23, para cada estadio de ERC, frente al aumento exagerado reflejo de una maladaptacion y que requiera la adopción de medidas terapéuticas.
Twenty years have passed since the identification of the klotho1 gene and the fibroblast growth factor 23 (FGF23) was discovered shortly after.2 The coincidence between the phenotypes of the mouse with FGF23 ablation and the klotho knock out mouse led to the recognition of klotho as a co-receptor for FGF233 and subsequent studies demonstrated the importance of klotho in phosphate homeostasis. This led to the fact that, despite FGF23 having been described in familial hypophosphatemic syndromes, successive studies on klotho and FGF23 are focused on chronic kidney disease (CKD), since the kidney is the main source of klotho and, at the same time, the main regulator of phosphate. The new information has revolutionized our previous perception of mineral homeostasis and place us in front of a new scenario in the clinical approach of the renal patient, in which FGF23 emerges as an early marker of phosphate retention and, at the same time, as a therapeutic target.
Aware of these new developments, in 2006, the kidney disease: improving global outcomes (KDIGO) working group introduced the term chronic kidney disease - mineral and bone disorders (CKD-MBD)4 to designate the alterations in mineral homeostasis that characterize CKD and limits the use of the term “renal osteodystrophy” to describe bone abnormalities caused by this imbalance. The purpose of this change is to highlight the precocity of these alterations which are initiated in early CKD and exclude the previous paradigm of late complication. Simultaneously, its scope is broadened by including the participation of other extra-osseous tissues with the appearance of soft tissue calcification, especially at the vascular level. Increased Phosphorus becomes a protagonist due to its key role in the development of vascular aging and calcification5 and it is starting to be considered toxic element above a narrow range and it cannot be subordinated the control of calcium by calciotropic hormones. In this scenario, the binomial FGF23/klotho turns out to be the missing piece in this complex puzzle that represents the endocrine regulation of mineral homeostasis.
FGF23 is protein produced by the bone that forms part of the subfamily of endocrine fibroblast factors (FGF). Although it shares the “core” common structure to all FGFs, FGF23 is differentiated by a characteristic cleavage segment between the amino acid sequence 176–179, surrounded by two arginine residues (Arg). After its transcription, which is regulated mainly by the chronic overload of phosphate, calcitriol and parathyroid hormone (PTH), FGF23 undergoes two other intra-osseous processes at the level of this segment: glycosylation mediated by the enzyme N-acetyl-galactosaminyl-transferase3 (GALNT3)6 that stabilizes the molecule it and limits a second degradation process regulated by proteases, especially by furins. Therefore, the circulating levels of FGF23 represent the balance between its transcription and its degradation. In blood it is detected the intact biologically active FGF23 molecule together with a series of inactive FGF23 fragments, consequence of the cleavage. At a physiological level, its target organs are those that express klotho: kidney, parathyroid and brain; a protein that forms binary complexes with the FGF1 receptor (FGFR1), increasing the affinity and specificity for FGF23. Thus, by signaling ras-mitogen-activated protein kinase (MAPK/RAS), the FGF23/klotho regulates phosphate by increasing phosphaturia by reducing tubular reabsorption of phosphate through the reduction of the NaPi2a and 2c co-transporters expression in the proximal tubular membrane. At the same time, it prevents phosphorus overload coming from the intestine or from bone resorption, through the suppressive action on vitamin D and PTH7 (Fig. 1A). Consequently calcium levels indirectly decrease and therefore, in situations of hypocalcemia which can be life-threatening, the body defends itself, inhibiting the synthesis of FGF23.8 At the level of the brain, its action is unknown, although it has been related to cognitive functions.
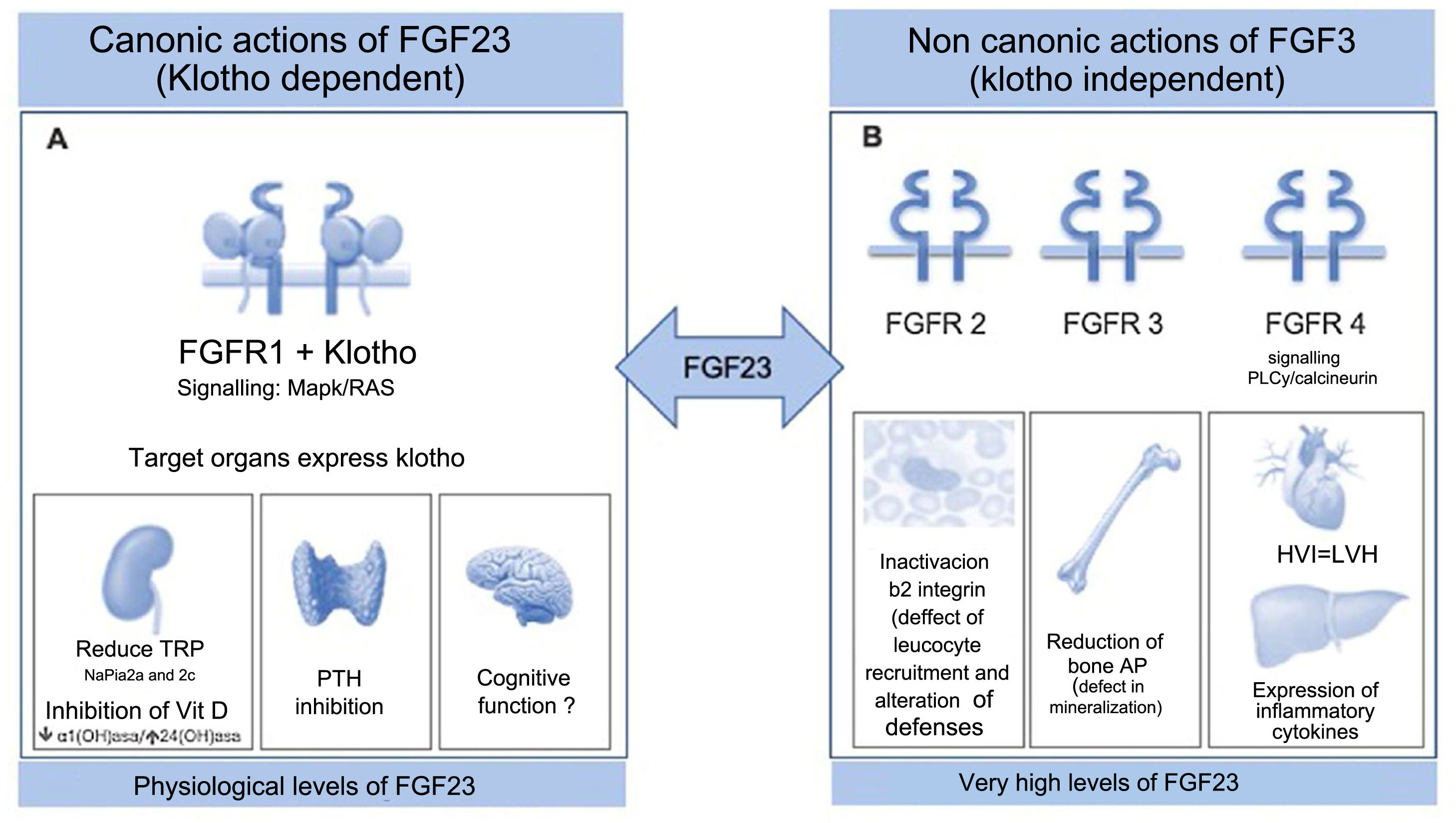
(A) Physiological actions requiring the expression of Klotho. FGF23 controls phosphorus homeostasis by binding to receptor 1 (FGFR1) with the participation of the protein Klotho which acts as a coreceptor, increasing sensitivity and specificity of the FGFR1 for FGF23; therefore its target organs are those expressing Klotho. The phosphaturic effect is mediated by the inhibition of sodium-phosphate cotransporter 2a and 2c; simultaneously, it inhibits the synthesis of calcitriol by suppressing the 1⍺-hydroxylase and promotes catabolism by 24-hydroxylase. This effect on vitamin D, together with the suppressive effect on PTH in the parathyroids, prevents new entry of phosphate into the extracellular space from the intestines and bone. Its action at the level of the brain is unknown, although it is related to cognitive functions.
(B) Toxic effects independent of Klotho. Extremely high levels of FGF23 that are evident in patients with the last stages of CKD, are able to activate other receptors in other organs that do not express Klotho causing important pathology. As an example, binding to FGFR2 deactivates the β2-integrin, altering leukocyte recruitment and the defense against the pathogens, or suppresses bone expression of alkaline phosphatase through activation of FGFR3, altering the mineralization as a result of pyrophosphate accumulation. FGF23 exerts a toxic effect on the myocardium through FGFR4 signalling, inducing left ventricular hypertrophy and on the liver it induces expression of inflammatory cytokines, worsening the inflammatory status which characterizes the renal patient and in turn it increases the synthesis of FGF23.
CKD: chronic kidney disease; FGF23: fibroblast growth factor 23; FGFR1: fibroblast growth factor receptor 1; FGFR2: fibroblast growth factor receptor t2; FGFR3: fibroblast growth factor receptor 3; FGFR4: fibroblast growth factor receptor 4; PTH: parathormone.
The development of methods to quantify circulating FGF23 made it possible to establish its involvement in CKD. The first enzyme immunoassays (EIA), not validated for clinical use and still existent, were classified into two types of format, depending on their antigenic configuration (Table 1). The format known as intact FGF23 (iFGF23), which exclusively recognizes the complete and active molecule of FGF23, better reflects its biological activity, although it is not equivalent, since the carboxy-terminal fragments also bind to the klotho coreceptor and, by competing with the iFGF23 reduce the signalling.9 Its main drawback is the effect of a possible ex vivo degradation by serum proteases, which is why plasma is the biological matrix of choice. At the same time, it shows greater intraindividual variability due to the influence of its circadian rhythm as FGF23, like any protein related to bone undergoes a nocturnal increase that culminates with a peak at around eight o'clock in the morning.10 By contrast, the carboxy-terminal EIA format (cFGF23), which recognizes both the active form and the C-terminal fragments, reflects the bone transcription of FGF23 and exhibits greater analytical stability. However, its interindividual variability is greater, making it difficult to establish biological intervals of reference. Both methods have advantages and disadvantages, it could even be interesting to quantify them together to identify the relationship between active FGF23 and its fragments. In any case, much of what we know today about CKD-MBD is due to the use of these EIAs, whose potential use in the clinical management of CKD offers important advantages over the classic biochemical markers of mineral metabolism.
Pre-analytical and analytical aspects in the measurement of FGF23.
| Pre-analytical aspects of FGF23 quantification |
|---|
| Circadian rhythm with peak secretion at 8 am Ideal extraction: 8−10 am |
| Possible in vitro degradation by proteases. Biological matrix: plasma |
| Analytical Aspects of FGF23 Quantification |
| Intact FGF23 method (iFGF23) | Method FGF23 C-Terminal (C-FGF23) |
|---|---|
| Exclusively measures intact form of the FGF23 | Intact form + carboxy-terminal fragments |
| Reflects transcription + intraosseous proteolysis | Reflects bone transcription exclusively |
| Represents the biological activity of FGF23 | Represents concentration, but not activity |
| Marked circadian rhythm | Little influenced by circadian rhythm |
| Major intra-individual variability | Major inter-individual variability |
| Low analytical stability ( in vitro degradation ) | Less affected by in vitro degradation |
The first advantage is how premature is its elevation. In CKD stage one it becomes evident the decrease in renal expression of Klotho,11 whose gene is very sensitive to inflammation and renal injury.12 As CKD progresses, together with the loss of functional nephrons, there is an inhibition in the expression of klotho due to the contribution of other factors, amongst them, the presence of uremic toxins that silence the klotho gene by hypermethylation,13 the increase in fractional excretion phosphorus,14 angiotensin II15 or vitamin D deficiency.16 Thus, as the kidney is the main source of klotho in the body, CKD represents the prototype of acquired klotho deficiency. The problem is that, the loss of nephrons and klotho, makes the failing kidney resistant to the action of FGF23 and the organism responds by increasing the bone synthesis of FGF23 to maintain normophosphatemia. This response begins in the early stages of CKD, with glomerular filtrations (GFR) of around 75 mL/min and the changes in FGF23 precedes the change in the rest of the biomarkers (Fig. 2). In fact, the highest values of FGF23 are predictive of secondary hyperparathyroidism (HPTs),17 which is what should be expected since logically, since secondary HPT results from the inhibition of vitamin D induced by FGF23 itself and its corresponding hypocalcemia. Along with this compensatory increase in FGF23, there are other factors that may modulate the bone expression of FGF23 in CKD. Parenteral iron supplementation induces FGF23 synthesis; an effect that is associated with some excipients such as sucrose or polymaltose,18 since it does not occur with all parenteral preparations or with the oral iron. By contrast, iron deficiency, anemia and hypoxic situations in general, also promote bone FGF23 transcript through the induction and stabilization of hypoxia induced factor 1 α (HIF1 α).19 Finally, the “inflammatory storm” that characterizes CKD is an important stimulus of bone expression of FGF23 by conditioning a functional iron deficiency. In this situation, the expression and stabilization of the HIF1α increases and, therefore, transcription of bone FGF23 augments; however, HIF1 α also increases simultaneously furins expression and the proteolysis of FGF23 to avoid unnecessary FGFR1signaling. This increase in FGF23 transcription promoted by inflammation could only be detected with the use of a cFGF23 method, which detects iFGF23 when it degrades into fragments. However, when the inflammatory process is chronic as in CKD, this compensatory proteolysis is exhausted and circulating levels of iFGF23 rise, this compensatory proteolytic mechanism that is altered in renal patients20 and therefore, in this situation, the two methods to quantify FGF23 have similar validity.
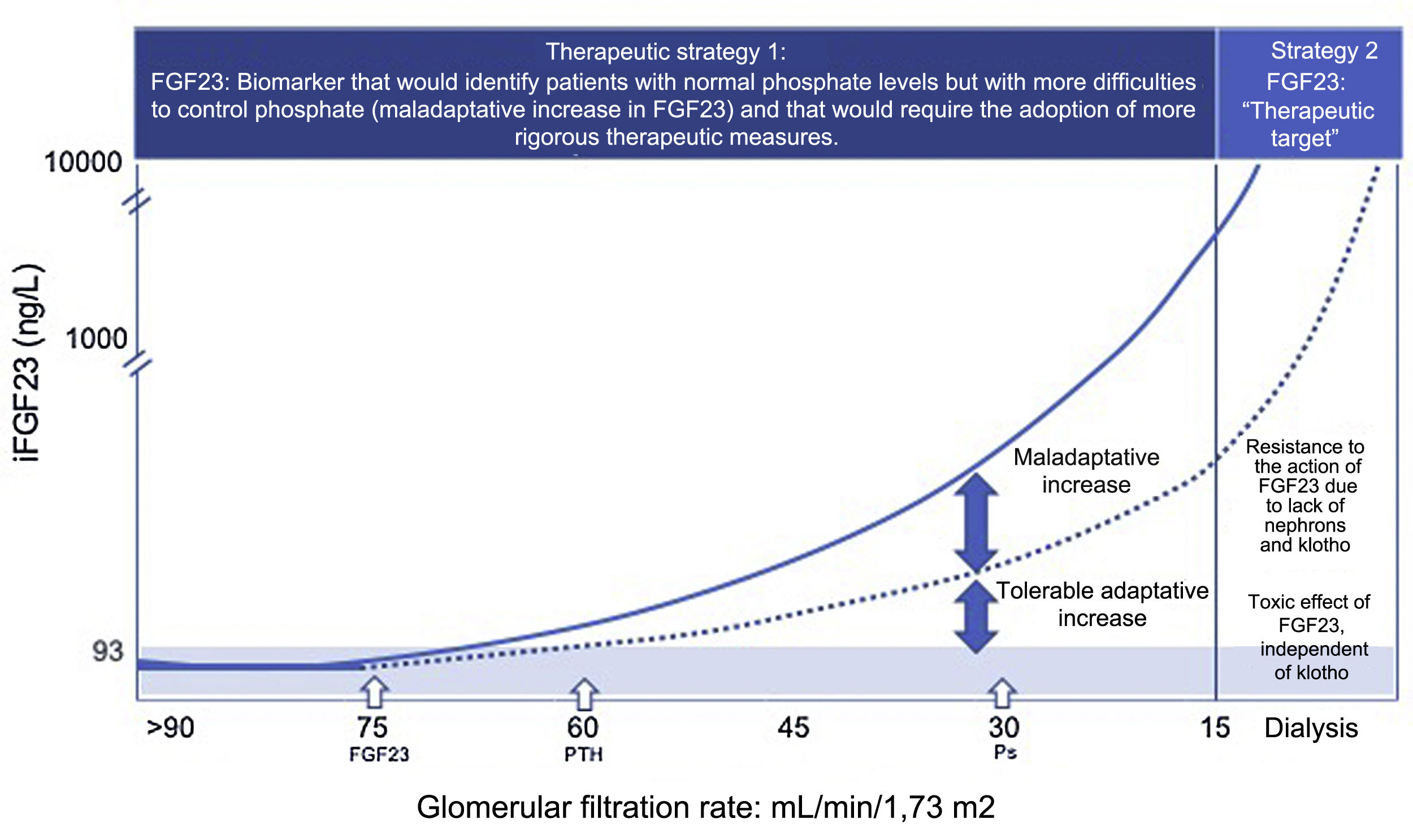
FGF23, a biomarker of early phosphate retention and a therapeutic target.
Therapeutic strategy 1: Faced with the loss of nephrons and klotho, an early adaptive increase in FGF23 occurs (eGFR: 75 mL/min/1.73 m2) to maintain normophosphatemia. When the compensatory phosphaturic capacity of FGF23 plus PTH is exceeded, hyperphosphatemia occurs which is usually observed with (eGFR: 30 mL/min/1.73 m2. and the vascular toxic effect of phosphate is present already.
The precocity of its elevation, together with its predictive value of mortality in conditionsof normophosphatemia, supports the usefulness of the quantification of FGF23, especially in the first stages of CKD, allowing to identify those normophosphatemic patients with a “maladaptive increase”, reflecting greater problems to control phosphorus, and that would require the adoption of more rigorous therapeutic measures.
Therapeutic strategy 2: In the final stages of CKD, the absence of nephrons and klotho generate resistance to the action of FGF23 and circulating levels of FGF23 are unlimited. These exaggerated values of FGF23 are capable of activating other FGFR receptors producing pathology in other organs and systems that do not express klotho. In this situation, FGF23 becomes a therapeutic target in an attempt to avoid toxicity.
CKD: chronic kidney disease; FGF23: fibroblast growth factor 23; FGFR: fibroblast growth factor receptor; PTH: parathyroid hormone.
As with other molecules, increased circulating levels of FGF23 are associated with faster progression of CKD.21 The marked hyperphosphatonisms, generated as a compensatory response to maintain normophosphatemia, reflect the difficulties to control phosphorus in even these early stages of CKD. At the same time, the greater phosphaturic effort induced by FGF23 has consequences on the nephron itself, and as mentioned before22 it may contribute to a greater loss in the expression of Klotho.14 Similarly, an exaggerated increased of FGF23 in CKD is associated with mortality.23 The advantage of this last relationship over the rest of the biomarkers is that, it increases as the multivariate analysis is applied, and it is significant in situations of normophosphatemia. And this is extremely important because phosphate is an insensitive marker, which only rises when the compensatory capacity of phosphaturic hormones (FGF23 and PTH) is exceeded (Fig. 2), a situation in which any therapeutic approach will be late because it will have already caused the initiation of processes of aging and vascular calcification. In this sense, FGF23 monitoring emerges as a valuable diagnostic and prognostic tool by allowing the identification of those patients who, even showing values of phosphataemia within the biological reference range, will have more problems controlling phosphorus homeostasis.
- 1
The contribution of FGF23 as an indicator of early phosphate retention implies a new orientation in patient treatment by offering us the possibility of being able to anticipate and timely control CKD-MBD, applying therapeutic strategies reserved for more advanced stages of CKD, such as non-calcium binders. FGF23 itself becomes a therapeutic target and the current objective of treatment would no longer be to correct the hyperphosphatemia that appears in advanced stages, but to guarantee adequate phosphate control from the initial stages of CKD, avoiding an increase in circulating FGF23 level beyond a supraphysiological adaptive range. Elevated levels of FGF23 imply a greater phosphaturic stress in the functioning nephrons, causing that a large amount of phosphate became concentrated in the tubular lumen. This precipitates with calcium and binds to fetuin-A, forming the so-called calciprotein particles, nanoparticles that, in turn, produce cytotoxicity in the tubular cell and more kidney damage.22 Very recent studies show an increase in these calciprotein particles dispersed in the blood of kidney patients, which would induce inflammatory response and vascular calcification which, after being deposited in bone, would activate the bone expression of FGF23.24 Therefore, the application of more restrictive dietary measures, with a favorable phosphorus/protein ratio to prevent poor nutrition, the use of non-calcium binders with advantages over the calcium binders in reducing FGF2325 and the control of other non-adaptive stimuli for FGf23 production such as anemia or IV administration of iron preparations with sucrose or polymaltose, at the moment in which this adaptive increase is exceeded. The idea is to minimize the impact of phosphate on the morbidity/mortality of the renal patient. The benefit of a correct control of CKD-MDB from the onset of CKD will condition the evolution of a future transplant, since the higher levels of FGF23 and PTH prior to this are predictive of persistent hyperphosphatasemia and tertiary hyperparathyroidism; situations that predispose to a greater loss of bone mass and an increased risk of graft loos and/or mortality.26,27
In the advanced CKD, when the number of nephrons and the amount of klotho is marginal, this adaptive mechanism is decompensated and there is resistance to the action of FGF23; furthermore, the presence of HPTs is an additional stimulus (Fig. 2). The circulating levels of FGF23 can reach more than 1000 times its biological reference range, which has a negative effects in the organism. Given the loss of klotho-dependent canonical signaling, the almost unlimited values of FGF23 activate receptors other than FGFR1, causing pathology in organs that do not express klotho (Fig. 1B); possibly, is facilitated by the presence of other proteins that would act as coreceptors. Thus, in the absence of the protective effect of klotho, FGF23 activates fibroblast growth factor receptor four (FGFR4) in the myocardium and, signaling via phospholipase C/calcineurin, increases cytoplasmic calcium levels and induces left ventricular hypertrophy (LVH).28,29 Interestingly, patients with X-linked familial hypophosphatemic rickets (XLH) do not present LVH despite increased FGF23; the reason for this discrepancy is probably due to the contribution of other factors, which are not present in the XLH phenotype, such as hyperphosphatemia and klotho deficiency, which favors the binding of FGF23 to FGFR4 and increases myocardial vulnerability due to the loss of the protective effects of soluble klotho on the cardiovascular system.30 This direct toxic effect of FGF23 on the myocardium is aggravated by other indirect effects of FGF23, such as the increase in the expression and renal activity of the sodium/chloride co-transporter (NCC), which by retaining sodium increases plasma volume and causes hypertensive damage,31 or a decrease in expression of angiotensin converting enzyme two (ACE2) and its counterregulatory beneficial effect on the renin angiotensin system (RAS).32 Through the activation of fibroblast growth factor receptor three (FGFR3), FGF23 exerts an autocrine suppressive action on bone transcription of non-tissue-specific alkaline phosphatase (TNAP) and, when pyrophosphate accumulates it is generated, a defect in mineralization.33 Excessive levels of FGF23 also activate fibroblast growth factor receptor two (FGFR2) on neutrophils, affecting β2-integrin, leukocyte recruitment, and host defense; a situation that justifies the greater susceptibility to infections shown by the renal patient.34 Apart from other effects, FGF23 also has an impact on the hepatocyte, since it expresses FGFR4 and its extra-canonical activation by FGF23 promotes the expression of inflammatory cytokines (IL6) and C-reactive protein (CRP), self-perpetuating inflammation and closing a vicious cycle.35 Of interest is the demonstration of ectopic production of FGF23 by liver cells of patients with autosomal dominant polycystic kidney disease, which explains the significantly higher values of plasma FGF23 shown in these cases in relation to other kidney patients with similar GFR.36
In advanced CKD, FGF23 remains a therapeutic target and the objective would be to minimize the effect of its toxicity on other organs and systems. Unlike the initial stages where the maintenance of circulating levels of FGF23 within its adaptive range prevails, reflecting an adequate control of phosphorus and CKD-MBD, in patients with CKD stage 5 the therapeutic strategy would be based on reducing its signaling, since the control of hyperphosphatonism is extremely complicated. A priori this may be surprising, since previous studies in experimental animal showed that the neutralization of FGF23 in CKD has benefits of hyperparathyroidism but at the cost of an increase in mortality37; a situation that is easily understood because by neutralizing FGF23, its important compensatory mechanism on CKD-MBD is blocked and the result is an uncontrolled increase in phosphate with the consequent toxic impact on vascular aging and mortality. For this reason, this therapeutic strategy is contraindicated as long as there is a functioning kidney on which FGF23 can act; however, in these final stages of CKD in which the adaptive phosphaturic mechanisms are no longer possible given almost total absence of klotho and nephrons, it would be reasonable to reduce this FGF23 non-canonical signaling, which only exerts harmful effects on the organism. This treatment hypothesis opens a new horizon to explore. Recently, a human monoclonal IgG1 antibody has been developed: burosumab, directed against the FGF23 molecule, indicated in XLH syndromes with very good and promising results.38 Another strategy could be the selective blocking of the receptors specifically involved in the adverse effects induced by FGF23. In this sense, the use of anti-FGFR4 antibodies could be another interesting approach to reduce inflammation, LVH, and morbidity/mortality in CKD.35
From salon nephrology to the patient’s bedsideAll of the previously explored supports the potential usefulness of FGF23 quantification in CKD. However, the first EIAs were not implemented in clinical laboratories because they were non-automated methods and not validated for clinical use. Currently, we have one of them. It is a chemiluminescent method (ECLIA) that quantifies iFGF23. There are several studies that validate its analytical behavior and evaluate it both in the general population and in patients with CKD.39,40 After several years of increased demand for a FGF23 method for clinical use, this automated ECLIA iFGF23 sets the starting point to individualize everything we know from epidemiological studies and, however, curiously, it is not fully implemented in daily practice. Its high price and the absence of clinical guidelines that recommend its quantification could be part of this problem, but two arguments must be taken into account. First, by definition, any method that is not automated is not contemplated in any clinical guideline and, therefore, the EIAs previously developed did not have a chance. Second, that the lack of experience and studies with this new method prevents establishing ranges and recommendations on its use in the context of the CKD-MBD. In summary, the current situation is that we have the ball in our court and it is time to work with this promising biomarker to answer many of the questions that we still ask ourselves and, once answered, we shall start using it properly in the management of CKD-MBD.
In the renal population, the previously mentioned validation studies39,40 demonstrate the expected inverse association between the increase in iFGF23 and GFR, and establish mean values and interquartile ranges for each stage of CKD. Logically, these ranges are higher than those observed in the general population since the elevation of FGF23 represents an adaptation mechanism of CKD-MBD. We can consider these ranges as an approximation; the problem is that they include both well-adapted and ill-adapted patients. Thus, the first objective that we propose would be to identify the added value for clinical decisions of each stage of CKD, that is, the specific threshold that delimits the expected physiological adaptive increase from the pathological exaggerated increase, in order to be able to discriminate the poorly controlled patients. This question is partially responded in a recently published study41 with a large cohort of patients with advanced CKD, which establishes a possible cut-off point for this automated ECLIA iFGF23, within the high range of FGF23 for each level of GFR, from which the risk of cardiovascular and renal events increases. Their results indicate that the optimal risk threshold for iFGF23 rises as the GFR declines and that the threshold for cardiovascular risk is: 177 ng/L for eGFR ≥ 30 mL/min; 228 ng/L for eGFR 20–29 mL/min and 528 ng/L for eGFR < 20 mL/min. For renal risk the threshold values are different: 141 ng/L for eGFR ≥ 30 mL/min; 192 ng/L for eGFR 20–29 mL/min and 311 ng/L for eGFR < 20 mL/min). This work represents the first attempt to address this challenge, offering new criteria for the clinical management of patients with advanced CKD and for future clinical trials. Nevertheless, fundamentally, it invites new studies that, in addition to endorsing its findings, consider other populations and other aspects that have not been evaluated.
Considering that the risk thresholds differ according to the type of event, it would be interesting to evaluate the association of this ECLIA with another type such as the development of HPTs or, especially, with vascular calcification. Likewise, although these cut-off points with a prognostic risk value are established for each stage of CKD, the early nature of the elevation of FGF23 makes interesting its evaluation in stages 3a and 3b of CKD; the idea is to anticipate the possible therapeutic strategies in patients at risk. Once these specific risk thresholds have been established, it will be necessary to demonstrate whether the correction of these aberrantly high values of iFGF23 provides benefits in relation to the morbidity/mortality of the renal patient. Let's take up this challenge to move from living room to patient bedside nephrology.
FinancingProject financed by the Health Research Fund ISC-III and co-financed with FEDER funds PI16/01298.
Conflict of interestsThe authors have no conflicts of interest to declare.
Please cite this article as: González-Casaus ML, Gonzalez-Parra E, Fernandez-Calle P, Buño-Soto A. FGF23: de la nefrología de salón a la cabecera del paciente. Nefrologia. 2021;41:276–283.



