


La nefropatía C1q es una enfermedad controvertida ya que algunos autores la consideran indistinguible de una nefropatía por cambios mínimos (ECM), mientras que otros la consideran una enfermedad de transición entre los cambios mínimos y la glomerulosclerosis segmentaria y focal (GEFS)1,2. Los criterios para el diagnóstico de la enfermedad incluyen depósitos predominantes de C1q en inmunofluorescencia y ninguna evidencia clínica o de laboratorio de lupus eritematoso sistémico3,4. Microscópicamente a diferencia de lupus, en la nefropatía C1q no debe existir enfermedad tubulointersticial5,6.
Presentamos un caso clínico de paciente de 21 años de edad, derivado a la consulta de nefrología desde pediatría en marzo de 2010, cuando tenía 14 años, por síndrome nefrótico córtico resistente. Previamente, en pediatría, diagnosticado de probable enfermedad de cambios mínimos se inició tratamiento con corticoides a dosis de 80mg/día.
La primera visita en nefrología revela una albumina de 2g/dl, proteínas totales 4,9g/dl, hemoglobina 15,9g/dl, creatinina 0,18mg/dl, iones no alterados, se observó una proteinuria de 4,9g en 24h, C3 y C4 162 y 22,9mg/g. La ecografía revela: ambos riñones aumentados de tamaño con aumento de parénquima renal en relación con el síndrome nefrótico. Se decide mantener el tratamiento esteroideo y es visto en consulta en enero del 2011 donde se aprecia aumento de la proteinuria pese al tratamiento. El estudio inmunológico es negativo para ANA, ANCA, ENA, DNA, anticuerpos anticardiolipina y anticuerpos MBG. Inmunoglobulinas: IgG 135mg/dl, resto de inmunoglobulinas dentro del rango. No se observaron datos de anemia y mantenía función renal normal. Se decide hacer biopsia renal (fig. 1): 30 glomérulos, con aumento leve de celularidad mesangial, no observándose engrosamientos de membranas básales, presencia de espinas (spikes), acúmulos de matriz ni colapsos capilares. Los componentes vascular y tubular no presentan alteraciones. En inmunofluorescencia directa se observó un depósito mesangial de C1q de forma intensa y en cuantía más escasa para C3 e IgG, siendo negativo para el resto de inmunoglobulinas, cadenas ligeras y fibrinógeno. Diagnóstico: enfermedad de cambios mínimos con depósito predominante de C1q, compatible con nefropatía C1q.
 Biopsia renal, morfológicamente se observa un leve aumento de la celularidad mesangial en algunos glomérulos. B) Estudio de inmunofluorescencia directa donde se observan depósitos de C1q.")
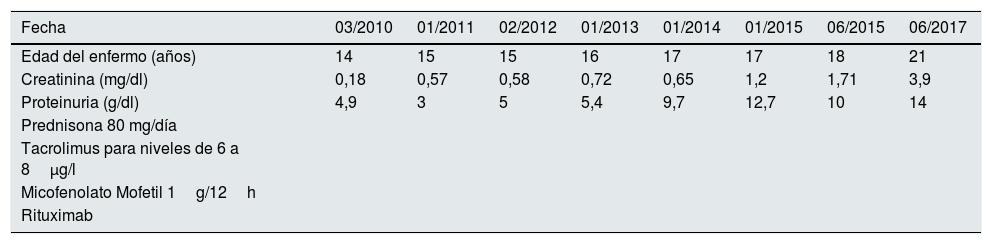
Se inicia tratamiento con tacrolimus para mantener niveles entre 6 y 8μg/l y se solicita estudio genético para descartar enfermedades relacionadas con el gen WT1 (síndrome de WAGR, Denis-Drash y síndrome de Fraiser), siendo todo negativo. Ante la falta de respuesta, en febrero del 2012 se inicia tratamiento con micofenolato mofetil en dosis crecientes hasta un máximo de 1g/12h, sin respuesta presentando en enero del 2015, una proteinuria de 9,4g/24h y deterioro de la función renal, estudio de autoinmunidad negativo (ANA, ANCA, DNA, ENA, MBG y crioglobulinas), por lo que se indica una segunda biopsia: 19 glomérulos, de los cuales un 50% (9) se encuentran completamente esclerosados, 6 no presentan alteraciones y los 4 restantes presentan grados variables de esclerosis, en 2 rodeando también la cápsula de Bowman. En la inmunofluorescencia no observaron depósitos de complemento, inmunoglobulinas, cadenas ligeras ni fibrinógeno en ninguna de las estructuras. A pesar de la desaparición de los depósitos de C1q en esta biopsia, el paciente presenta 5 años de tratamiento con un declive progresivo de su función renal por lo que se decide iniciar una cuarta línea de tratamiento con rituximab, recibiendo 4 bolus semanales en junio de 2015 sin mejoría de la proteinuria e importante deterioro de la función renal (tabla 1), agotando las posibles líneas de tratamiento y derivando el paciente a ERCA.
Relación entre tratamiento, proteinuria y función renal desde marzo de 2017 hasta julio de 2017 en paciente con diagnóstico de nefropatía C1q
| Fecha | 03/2010 | 01/2011 | 02/2012 | 01/2013 | 01/2014 | 01/2015 | 06/2015 | 06/2017 |
|---|---|---|---|---|---|---|---|---|
| Edad del enfermo (años) | 14 | 15 | 15 | 16 | 17 | 17 | 18 | 21 |
| Creatinina (mg/dl) | 0,18 | 0,57 | 0,58 | 0,72 | 0,65 | 1,2 | 1,71 | 3,9 |
| Proteinuria (g/dl) | 4,9 | 3 | 5 | 5,4 | 9,7 | 12,7 | 10 | 14 |
| Prednisona 80 mg/día | ||||||||
| Tacrolimus para niveles de 6 a 8μg/l | ||||||||
| Micofenolato Mofetil 1g/12h | ||||||||
| Rituximab |
El paciente recibió a lo largo del tiempo dependiendo del estadio de la enfermedad renal tratamiento con: ácido acetilsalicílico, hidroferol 0,266μg, estatinas, inhibidor de la bomba de protones, quelantes de fosforo y antihipertensivos.
Los anticuerpos anti-C1q están dirigidos contra el primer componente del complemento, son en su mayoría, del subtipo IgG predominantemente IgG1 e IgG2 demostrando una fuerte correlación entre su presencia y el daño renal. Los mecanismos por los que se encuentra C1q en las biopsias renales serían: 1) que vaya unido a la porción Fc de las Ig implantadas o de inmunocomplejos circulantes; 2) restos apoptóticos podrían captar C1q facilitando su aclaramiento; 3) el C1q podría unirse a la proteína C reactiva, amiloide o Ig atrapadas en el glomérulo; 4) unión directa del C1q en forma específica con células del parénquima renal; 5) atrapamiento pasivo, y 6) reacción cruzada con antígenos similares al C1q7–10. De este modo se explica que cada vez que estamos frente a una nefropatía con intervención de la vía clásica del complemento se puedan observar depósitos de C1q en la biopsia.
Por lo tanto, los pacientes que tienen nefropatía C1q y GEFS tienen más probabilidades de progresar a enfermedad renal terminal. No hay ensayos aleatorizados que hayan evaluado el tratamiento de la nefropatía C1q. La terapia implica el tratamiento de la lesión microscópica subyacente.



