


El diagnóstico de certeza de algunas enfermedades renales solo es posible con un estudio genético. Es el caso de las nefropatías túbulo-intersticiales autosómicas dominantes (NTIAD), cuyo término fue establecido por las guías KDIGO en el año 20151.
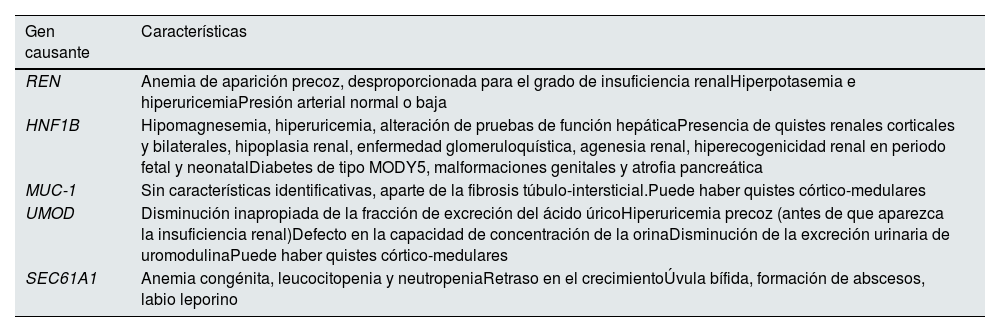
Se manifiestan con una pérdida progresiva de función renal, con proteinuria negativa o anodina y generalmente con sedimento urinario normal. En la ecografía, los riñones son de tamaño normal o pequeño, con presencia inconstante de quistes corticomedulares. La biopsia renal es inespecífica, dado que solo muestra datos de fibrosis intersticial y atrofia tubular. Los genes causantes conocidos más frecuentes son 5: UMOD, MUC-1, REN, HNF1B y SEC61A1, con características clínicas diferenciales entre ellos (tabla 1)2-5. La penetrancia es cercana al 100% y puede existir variabilidad intra- e interfamiliar. Constituyen el tercer grupo de enfermedad renal monogénica, después de la poliquistosis renal autosómica dominante y la enfermedad del colágeno de tipo iv4.
Características de las diferentes NTIAD
| Gen causante | Características |
|---|---|
| REN | Anemia de aparición precoz, desproporcionada para el grado de insuficiencia renalHiperpotasemia e hiperuricemiaPresión arterial normal o baja |
| HNF1B | Hipomagnesemia, hiperuricemia, alteración de pruebas de función hepáticaPresencia de quistes renales corticales y bilaterales, hipoplasia renal, enfermedad glomeruloquística, agenesia renal, hiperecogenicidad renal en periodo fetal y neonatalDiabetes de tipo MODY5, malformaciones genitales y atrofia pancreática |
| MUC-1 | Sin características identificativas, aparte de la fibrosis túbulo-intersticial.Puede haber quistes córtico-medulares |
| UMOD | Disminución inapropiada de la fracción de excreción del ácido úricoHiperuricemia precoz (antes de que aparezca la insuficiencia renal)Defecto en la capacidad de concentración de la orinaDisminución de la excreción urinaria de uromodulinaPuede haber quistes córtico-medulares |
| SEC61A1 | Anemia congénita, leucocitopenia y neutropeniaRetraso en el crecimientoÚvula bífida, formación de abscesos, labio leporino |
Presentamos 2casos diagnosticados en nuestro centro.
El primer caso es una paciente de 23 años, sin antecedentes médicos de interés, que fue remitida por deterioro de función renal con creatinina de 1,4mg/dl, CKDEPI 53ml/min/1,73 m2, cociente de albúmina/creatinina en orina de 7,4mg/g y sin alteraciones en el sedimento urinario. En la ecografía, los riñones eran de tamaño y morfología normales, aunque levemente hiperecogénicos, sin evidencia de quistes renales. No presentaba nada destacable en la anamnesis: no tenía historia de infecciones de orina ni de cólicos nefríticos, sin ingesta de nefrotóxicos, ni factores de riesgo cardiovascular (presentaba cifras de presón arterial de 120/70mmHg). Como antecedentes familiares (fig. 1): su bisabuela materna falleció a los 35 años de «nefropatía», su abuela materna inició diálisis a los 45 años, su tía materna inició diálisis a los 55 años y su madre inició diálisis a los 48 años. En los 3casos, la enfermedad renal crónica (ERC) no estaba filiada y se había relacionado con perfil vascular por presentar datos de hipertensión arterial a los 35-40 años. Ampliamos estudio (ANA, anti-ADN, ANCA, ENA, C3/C4, IgA/M/G, proteinograma, VIH/VHB/VHC): todo resultó negativo o normal. La paciente presentó un deterioro progresivo de la función renal sin otras causas atribuibles. Rechazó una biopsia renal que se le propuso. Ante el perfil autosómico dominante familiar, solicitamos un estudio genético, en el que se detectó una variante patogénica en el gen MUC-1, causante de nefropatía túbulo-intersticial autosómica dominante. Actualmente la paciente tiene 28 años, con enfermedad renal crónica avanzada (creatinina 5,2mg/dl CKDEPI 10ml/min/1,73 m2), por lo que la progresión ha sido más rápida que en sus otros familiares.

El segundo caso se trata de un varón de 29 años, con antecedentes de interés de hipertensión arterial de un año de evolución con buen control con un fármaco a dosis bajas, meningitis (2015) y apendicectomía. Fue remitido a nuestra consulta por hallazgo de deterioro de función renal (creatinina 2mg/dl, CKDEPI 38ml/min/1,73 m2), con cociente de albúmina/creatinina en orina de 290mg/g, sin alteraciones en el sedimento urinario. No disponíamos de analíticas ni de informes previos (antes residía en otra ciudad). Ecográficamente los riñones medían 10cm y presentaban mala diferenciación corticomedular y pequeños quistes corticales bilaterales. Como antecedentes familiares: su abuelo paterno inició diálisis a los 73 años (no disponían de informes, residía en otra ciudad y ya había fallecido), padres sin antecedentes de enfermedad renal y sin ningún otro antecedente nefrológico conocido en su familia. No había sordera en la familia. Realizamos un estudio amplio (ANA, anti-ADN, ANCA, ENA, C3/C4, crioglobulinas, IgA/M/G/IgG4, proteinograma, VIH/VHB/VHC): todo fue negativo o normal. Un estudio de despistaje de Fabry (niveles de α-galactosidasa) también fue negativo. No planteamos realizar una biopsia renal por los datos importantes de cronicidad en la ecografía. Ante una ERC no filiada, la edad del paciente y el antecedente familiar, decidimos solicitar estudio genético, que detectó una variante patogénica en el gen MUC-1, causante de nefropatía intersticial autosómica dominante.
Como conclusión, se debe sospechar una NTIAD en pacientes jóvenes, con una ERC no filiada sin datos de glomerulonefritis y con historia familiar de nefropatía, cuyo diagnóstico de certeza únicamente es posible con un estudio genético6,7. Gracias al avance tecnológico de los estudios genéticos en los últimos años, en la actualidad son pruebas asequibles y eficientes para pacientes bien seleccionados8,9.



