

Confirming a diagnosis for certain renal diseases is only possible with a genetic study. This is the case for autosomal dominant tubulointerstitial kidney disease (ADTKD), as defined by the KDIGO guidelines in 2015.1
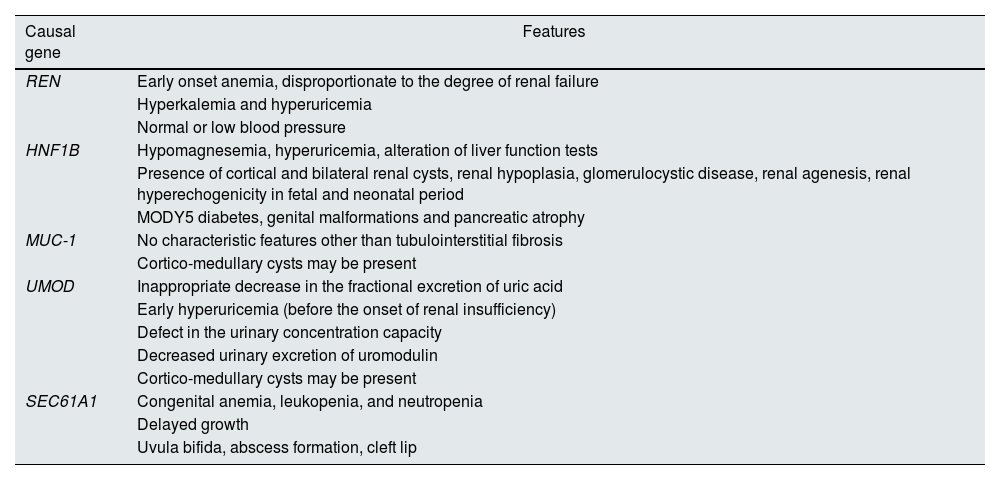
ADTKDs manifest as progressive loss of renal function, with negative or anodyne proteinuria and usually with normal urinary sediment. On ultrasound, the kidneys are normal or small size, with inconsistent presence of corticomedullary cysts. Renal biopsy is nonspecific, showing only evidence of interstitial fibrosis and tubular atrophy. The five most frequent known causative genes are: UMOD, MUC-1, REN, HNF1B and SEC61A1, with differential clinical characteristics among them (Table 1).2–5 Penetrance is close to 100% and there may be intra- and interfamilial variability. They constitute the third most common group of monogenic renal disease, after autosomal dominant polycystic kidney disease and type IV collagen disease.4
Characteristics of the different NTIADs.
| Causal gene | Features |
|---|---|
| REN | Early onset anemia, disproportionate to the degree of renal failure |
| Hyperkalemia and hyperuricemia | |
| Normal or low blood pressure | |
| HNF1B | Hypomagnesemia, hyperuricemia, alteration of liver function tests |
| Presence of cortical and bilateral renal cysts, renal hypoplasia, glomerulocystic disease, renal agenesis, renal hyperechogenicity in fetal and neonatal period | |
| MODY5 diabetes, genital malformations and pancreatic atrophy | |
| MUC-1 | No characteristic features other than tubulointerstitial fibrosis |
| Cortico-medullary cysts may be present | |
| UMOD | Inappropriate decrease in the fractional excretion of uric acid |
| Early hyperuricemia (before the onset of renal insufficiency) | |
| Defect in the urinary concentration capacity | |
| Decreased urinary excretion of uromodulin | |
| Cortico-medullary cysts may be present | |
| SEC61A1 | Congenital anemia, leukopenia, and neutropenia |
| Delayed growth | |
| Uvula bifida, abscess formation, cleft lip |
We present 2 cases diagnosed in our center.
The first case is a 23-year-old female patient, with no relevant medical history, who was consulted for renal function deterioration with creatinine of 1.4 mg/dl, CKDEPI 53 ml/min/1.73 m2, urine albumin/creatinine ratio of 7.4 mg/g and no alterations in urinary sediment. On ultrasound, the kidneys were of normal size and morphology, although slightly hyperechogenic, with no evidence of renal cysts. There was nothing remarkable in the anamnesis: she had no history of urinary tract infections or nephritic colic, no nephrotoxic intake and no cardiovascular risk factors (she had blood pressure of 120/70 mmHg). Regarding family history (Fig. 1): her maternal great-grandmother died at 35 years of age of “nephropathy,” her maternal grandmother started dialysis at 45 years of age, her maternal aunt started dialysis at 55 years of age and her mother started dialysis at 48 years of age. In all 3 cases, chronic kidney disease (CKD) was not affiliated and had been related to vascular profile because she had arterial hypertension at 35–40 years of age. We performed more studies (ANA, anti-DNA, ANCA, ENA, C3/C4, IgA/M/G, proteinogram, HIV/HBV/HCV): all were negative or normal. The patient presented progressive deterioration of renal function with no other potential causes. She refused renal biopsy. Given the familial autosomal dominant profile, clinicians requested a genetic study, which detected a pathogenic variant in the MUC-1 gene, causing NTIAD. The patient is now 28 years old, with advanced chronic kidney disease (creatinine 5.2 mg/dl CKDEPI 10 ml/min/1.73 m2), so the progression has been faster than in her other relatives.

The second case is a 29-year-old male, with medical history of arterial hypertension of one year with good control with low-dose antihypertensive drugs; he also had meningitis (2015) and previous appendectomy. He was referred to us because of renal function deterioration (creatinine 2 mg/dl, CKDEPI 38 ml/min/1.73 m2), with an urine albumin/creatinine ratio of 290 mg/g, with no alterations in urinary sediment. We had no previous lab analyses or reports (he used to live in another city). Ultrasound revealed the kidneys measured 10 cm and presented poor corticomedullary differentiation and small bilateral cortical cysts. As for family history: his paternal grandfather started dialysis at age 73 (no reports were available, he resided in another city and he was deceased), parents had no history of renal disease and no other known nephrological history in his family. There was no deafness in the family. We performed an extensive study (ANA, anti-DNA, ANCA, ENA, C3/C4, cryoglobulins, IgA/M/G/IgG4, proteinogram, HIV/HBV/HCV): everything was negative or normal. A Fabry screening study (α-galactosidase levels) was also negative. We did not consider performing a renal biopsy due to the important chronicity data in the ultrasound. In view of the non-filial CKD, the patient’s age and the family history, we decided to request a genetic study, which detected a pathogenic variant in the MUC-1 gene, that was the cause of autosomal dominant interstitial nephropathy.
In conclusion, NTIAD should be suspected in young patients with non-infiltrative CKD without glomerulonephritis data and with a family history of nephropathy, whose reliable diagnosis is only possible with a genetic study.6,7 Thanks to technological advances in genetic studies in recent years, there are now affordable and efficient tests for well-selected patients.8,9



