





Presentamos el caso de una paciente de 43 años con un cuadro de síndrome nefrítico en el contexto de una glomerulonefritis extracapilar tipo I. La presentación, la disociación clínico-histológica evidente y la respuesta al tratamiento inmunosupresor han sido muy peculiares.
INTRODUCCIÓN
La enfermedad por anticuerpos por antimembrana basal glomerular (anticuerpos anti-MBG) es una entidad con afectación renal, autoinmune, agresiva y relativamente poco frecuente en nuestro medio. Su presentación clínica habitual es una insuficiencia renal rápidamente progresiva con hematuria y proteinuria. Presenta un mal pronóstico renal a pesar del tratamiento combinado con recambio plasmático e inmunosupresores.
Presentamos una paciente de 43 años con un cuadro de síndrome nefrítico en el contexto de una glomerulonefritis extracapilar tipo I. La presentación, la disociación clínico-histológica y la respuesta al tratamiento han sido muy peculiares.
CASO CLÍNICO
Paciente de 43 años, sin alergias medicamentosas conocidas ni antecedentes médicos de interés, que es derivada a nuestro centro por presentar episodio de epigastralgia con malestar general de 15 días de evolución y algún vómito autolimitado. No presenta anorexia ni otra sintomatología.
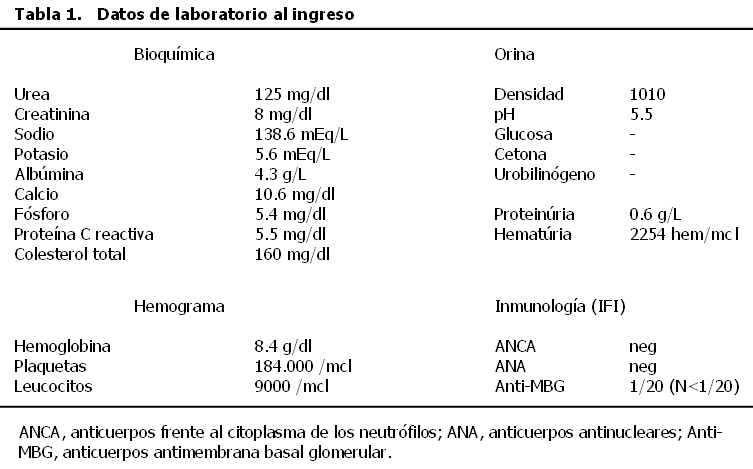
En la exploración física está normotensa, destaca una ligera palidez mucocutánea, tonos cardíacos rítmicos, sin soplos, sin edemas periféricos, tórax y abdomen sin hallazgos. No presenta lesiones cutáneas. En los datos de laboratorio al ingreso (tabla 1) destaca una anemia normocrómica normocítica y un síndrome nefrítico con microhematuria y proteinuria de 0,6 g/día. En el estudio inmunológico destaca únicamente un título borderline de anticuerpos antimembrana basal glomerular, medido por inmunofluorescencia indirecta (IFI). Las serologías virales (virus de la hepatitis B y C, virus de la inmunodeficiencia humana) y el resto de los anticuerpos fueron negativos.
Se realiza una ecografía renal que muestra unos riñones de tamaño normal (130 x 50 mm), ecogenidad cortical aumentada, sin imágenes de dilatación de vía urinaria. Radiografía de tórax normal.
Ante la agresividad clínica del cuadro se inicia tratamiento con tres bolos de 1 g/día de metilprednisolona y posteriormente a dosis de 1 mg/kg/día con pauta descendente según protocolo, previos a la espera del resultado de la biopsia renal. Se instaura tratamiento con bolos de ciclofosfamida intravenosa mensual a dosis de 300 mg, ajustados a su peso y función renal, hasta completar siete bolos. Se instaura tratamiento profiláctico con sulfametoxazol/trimetroprim. Se obtiene el resultado de la biopsia renal, que comprende 16 glomérulos. En cinco de ellos se evidencian lesiones proliferativas y necrosantes, con rotura de la pared de los capilares glomerulares y formación de pequeñas semilunas celulares (figura 1). El resto de los glomérulos fueron normales. El intersticio presenta un ligero infiltrado linfocitario de predominio periglomerular. Algunos túbulos muestran vacuolización del epitelio tubular sin claros signos de necrosis tubular aguda; en otros se evidencia la luz ocupada por cilindros granulares, hialinos y hematíes. El estudio de inmunofluorescencia directa presenta una positividad lineal en la pared de los capilares glomeurales para IgG (figura 2), kappa y lambda. El resto de cadenas pesadas y C3 son negativos.
Ante los hallazgos de IgG lineal en la immunofluorescencia, se decide realizar tratamiento con sesiones de plasmaféresis. Se completan cinco sesiones de plasmaféresis, con recambio plasmático de 3 litros y reposición de seroalbúmina al 4%. Sin respuesta clínica y a pesar de mantener diuresis durante todo el proceso, la paciente requiere tratamiento renal sustitutivo con hemodiálisis mediante catéter yugular derecho e inicio de tratamiento con eritropoyetina. A los 3 meses del inicio del cuadro y ante la persistencia del fracaso renal, se coloca un catéter tipo Tenchkoff mediante cirugía laparoscópica, iniciando posteriormente diálisis peritoneal. Realiza pauta de CAPD durante un período de 6 meses.
Al noveno mes del inicio del cuadro se objetiva una mejoría del filtrado glomerular, permitiendo retirar a la paciente del tratamiento renal sustitutivo. A los 12 meses, sin tratamiento médico, la paciente presenta un filtrado glomerular de 37 ml/min con sedimento de orina negativo. Dada la evolución favorable de la función renal, la disociación clínico-patológica inicial y los títulos borderline de los anticuerpos anti-MBG, para determinar los signos de actividad de la enfermedad se decide realizar una nueva determinación por IFI de los anticuerpos antimembrana basal glomerular, que resultan negativos, y una nueva biopsia renal. Se obtienen 12 glomérulos, cuatro de los cuales muestran lesiones de esclerosis segmentaria con formación de sinequias en la cápsula de Bowman, sin observarse lesiones proliferativas ni necrosantes (figura 3). El intersticio muestra pequeños focos de infiltrado linfocitario, fibrosis y atrofia tubular. La inmunofluorescencia es negativa. No se observan signos de actividad de la enfermedad basal. En la figura 4 se expresa la evolución clínica y los distintos tratamientos recibidos.
DISCUSIÓN
La enfermedad por anticuerpos anti-MBG es una enfermedad autoinmune caracterizada por la presencia en el suero de autoanticuerpos IgG dirigidos contra un antígeno localizado en la cadena alfa-3 del colágeno tipo IV, presente principalmente en la membrana basal glomerular (MBG).
La enfermedad por anticuerpos anti-MBG habitualmente se manifiesta con un cuadro clínico de insuficiencia renal rápidamente progresiva (RP), con un sustrato histológico en el que predomina la proliferación extracapilar con depósitos lineales de IgG y C3 a lo largo de la MBG (GNRP tipo I). Más de la mitad de los enfermos presentan afectación renal y hemorragia alveolar concomitante (síndrome de Goodpasture).
La enfermedad por anticuerpos anti-MBG es relativamente poco frecuente, pero con amplia distribución en el mundo. Su incidencia se desconoce, aunque se han descrito numerosos casos. En nuestro medio, la incidencia de la glomerulonefritis extracapilar ha ido decreciendo en los últimos años, siendo la tipo III la más frecuente de la GNRP (65,1%), seguida de la tipo II (20%) y la tipo I con un 14,6%1. Tiene una incidencia bimodal, con pico en la tercera década y entre la sexta y séptima décadas de la vida, con una ligera prevalencia del sexo masculino.
La presencia de anticuerpos anticitoplasma de neutrófilo (ANCA) se ha descrito en una tercera parte de los casos de esta enfermedad, con una mayor prevalencia de la especificidad antimieloperoxidasa (MPO). Algunos autores sugirieren el efecto potencialmente beneficioso de la coexistencia de ANCA y AC anti-MB en cuanto a mejor pronóstico, comparado con aquellos casos en los que únicamente presentaban positividad para AC anti-MBG2,3.
La información que proporciona la biopsia renal es crucial tanto para el diagnóstico como para el pronóstico. En la microscopia óptica las semilunas son la expresión morfológica de la proliferación de células parietales en la cápsula de Bowman. En la GN extracapilar tipo I estas semilunas se encuentran normalmente en el mismo estadio evolutivo, lo que indica una agresión glomerular aguda generalizada. La inmunofluorescencia es esencial, con la presencia típica de depósitos lineales de IgG, y ocasionalmente (10-15%) también de IgA o IgM, así como de complemento, a lo largo de la membrana basal de todos los glomérulos4. En nuestra paciente, sin embargo, sorprende la disociación clínico-patológica. Con la presencia de semilunas en menos del 50% de los glomérulos y sin signos de lesión tubular consistentes, perdió la función renal y requirió el inicio de tratamiento renal sustitutivo durante 9 meses. Es difícil encontrar una clara explicación para esta disociación tan evidente y no encontramos en la bibliografía referencias semejantes. A pesar de ello, la baja agresividad histológica concuerda con la apreciable recuperación de la función renal posterior.
El recambio plasmático o plasmaféresis a fin de eliminar o reducir los anticuerpos circulantes5,6, la inmunosupresión con ciclofosfamida y los corticoides para frenar y modular la síntesis de anticuerpos son los principales pilares del tratamiento de la enfermedad por anticuerpos anti-MB7,8. La supervivencia renal al año del diagnóstico es baja8,9 y la respuesta al tratamiento depende de las cifras de creatinina en el momento del diagnóstico. Nuestra paciente tenía elementos de pronóstico diversos para responder a un tratamiento agresivo convencional con prednisona en bolos, plasmaféresis y ciclofosfamida ajustada a la función renal. Por un lado presentaba insuficiencia renal avanzada en el momento de la presentación, hecho que reduce las posibilidades de respuesta al tratamiento6,10. Por otro lado, mantuvo la diuresis conservada a lo largo de los 9 meses iniciales y la lesión histológica era poco agresiva, signos que favorecen una buena respuesta al tratamiento6,7. Con esta constelación de signos y síntomas clínicos e histológicos, presentó un cuadro clínico muy peculiar: ausencia de respuesta inicial y, finalizado el tratamiento, respuesta tardía progresiva y negativización de los signos de actividad de la enfermedad (ausencia de hematuria, desaparición de los depósitos lineales de IgG y de los anticuerpos anti-MBG circulantes y mejoría progresiva de la función renal).
Casos clínicos como el que presentamos refuerzan las actitudes terapéuticas agresivas ante nefropatías glomerulares proliferativas agudas que, a pesar de cursos clínicos de evolución tórpida, presentan signos de buen pronóstico clínicos o histológicos.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.

Tabla 1. Datos de laboratorio al ingreso

Figura 1. Tinción de tricrómico de Masson. Glómerulo con lesiones proliferativas-necrosantes. Semiluna celular. Intersticio con infiltrado linfocitario periglomerular (x 600).

Figura 2. Inmunofluorescencia. Presencia de depósitos IgG lineales a lo largo de la membrana basal glomerular (x 600).

Figura 3. Tinción de hematoxilina-eosina. Esclerosis segmentaria con formación de sinequias. Intersticio con infiltrado linfocitario. Fibrosis y atrofia tubular (x 400).

Figura 4. Evolución clínica de la paciente.


