





La afectación renal asociada a linfoma es un fenómeno conocido pero frecuentemente no caracterizado debido a la baja frecuencia con que se realizan biopsias en estos pacientes. Varios patrones histológicos pueden coexistir y pasar desapercibidos sin un estudio histopatológico. La infiltración parenquimatosa renal por linfoma no es infrecuente, y se ha encontrado hasta en un 34% (post mortem) y 14% (pre mortem), aunque tiene una baja incidencia de manifestaciones clínicas. Existen diferentes patrones de lesión renal asociados a linfoma y destaca la asociación de enfermedad de cambios mínimos con linfoma de Hodgkin. La afectación renal asociada a paraproteínas sintetizadas por un linfoma linfoplasmocitario es una asociación excepcional pese a que existen un 20% de pacientes afectados por dichos linfomas que presentan crioglobulinemia. En la literatura se han publicado casos de enfermedad de cadenas ligeras, amiloidosis, glomerulonefritis inmunotactoide como causas de paraproteinemia, proteinuria e insuficiencia renal en pacientes con linfoma. Presentamos un caso de asociación entre paraproteinemia, glomerulonefritis membrano-proliferativa y la aparición clínicamente evidente de un linfoma linfoplasmocitario en ausencia de infección por virus de la hepatitis C. Esto demuestra la afectación polimorfa que pueden presentar los linfomas en el riñón y el valor de la nefropatología en el diagnóstico y pronóstico de las enfermedades hematológicas que cursan con paraproteinemia.
Kidney involvement associated to lymphoma is a known phenomenon but frequently not characterized due to the low frequency with which biopsies are realized in these patients. Several histological patterns can co-exist and happen unnoticed without a biopsy. Parenchyma infiltration in kidney for lymphoma has been found in 34% (post-mortem) and 14% (pre-mortem) and have low incident of clinical manifestations. Other patterns of renal injury are associated to lymphomaand minimal changes disease is especially related with Hodgkin’s lymphoma. Renal lesions associated to paraprotein in lymphoplasmocitic lymphoma are an exceptional association, in spite of in 20% of them, appear cryoglobulinemia. There are a few cases reported in the literature with different histological patterns: light-chain disease, amyloidosis, and immuotactoid glomerulopathy related with kidney injury in patients with lymphoma. A 39-year-old male presented an association among paraproteinemia, membrano-proliferative glomerulonephritis no hepatitis C virus related and lymphoplasmocitic lymphoma with renal infiltration. This case emphasized the variety of renal lesions that lymphomas could trigger and the value of the nephropatology in the diagnosis and outcome of the hematologic diseases with paraproteinemia.
INTRODUCCIÓN
La afectación renal asociada a linfoma es un fenómeno conocido pero frecuentemente no caracterizado debido a la baja frecuencia con que se realizan biopsias en estos pacientes, en quienes varios patrones histológicos pueden coexistir y pasar desapercibidos sin un estudio histopatológico1.
La infiltración parenquimatosa renal por linfoma no es un fenómeno infrecuente, y en estudios post mortem se ha encontrado hasta en el 34% de los casos estudiados, pero sólo el 14% de estos casos fueron diagnosticados pre mortem debido a la baja incidencia de manifestaciones clínicas y/o insuficiencia renal en estos pacientes. Además, en diferentes series se ha demostrado que la infiltración linfomatosa renal se asocia con un pobre pronóstico desde el punto de vista hematológico2.
Sin embargo, existen otros patrones de lesión renal asociados con linfomas, entre los que destaca la asociación de la enfermedad de cambios mínimos con el linfoma de Hodgkin, en la que esta glomerulopatía representa el 40% de los casos con patología renal3. Por el contrario, la asociación de glomerulonefritis membranosa y linfoma es mucho menos frecuente que la publicada para esta enfermedad glomerular en relación con tumores de órgano sólido1. La afectación renal asociada a paraproteínas sintetizadas por el linfoma linfoplasmocitario es una asociación excepcional; a pesar de que aproximadamente un 20% de los linfomas linfoplasmocitarios cursan con crioglobulinemia y a la casi siempre presente presencia de gammapatía monoclonal IgM kappa4. En la literatura se han comunicado casos de enfermedad de cadenas ligeras5, amiloidosis5, o glomerulonefritis inmunotactoide6 como causas de proteinuria e insuficiencia renal en pacientes con linfoma. A continuación presentamos un ejemplo de asociación entre la aparición en orden cronológico de paraproteinemia, glomerulonefritis membrano-proliferativa y la aparición clínicamente evidente de un linfoma linfoplasmocitario (LLP) en ausencia de infección por virus de hepatitis C (VHC), que demuestra la afectación polimorfa que pueden presentar los linfomas en el riñón y el valor de la nefropatología en el diagnóstico y en el pronóstico de la enfermedad hematológica que cursa con paraproteinemia.
CASO CLÍNICO
Hombre de 39 años sin hábitos tóxicos, alérgico al ácido acetilsalicílico (AAS) y al diclofenaco. Consulta inicialmente por tumefacción de partes blandas asociada a púrpura palpable en ambas extremidades inferiores en abril de 2001. Se realizó estudio inmunológico que puso de manifiesto crioglobulinemia positiva con criocrito de 9,2% (IgM kappa de carácter monoclonal y componente IgG). Las serologías para virus hepatotropos y virus de la inmunodeficiencia humana (VIH) fueron negativas, así como también dieron negativos los anticuerpos antifosfolipídicos.
En la exploración física destacaba la presencia de púrpura palpable en las extremidades inferiores; el resto del examen fue anodino. Se realizó una tomografía computarizada (TC) toracoabdominal en la que no se evidenciaron adenopatías ni visceromegalias. Con la orientación de una vasculitis cutánea, se inició tratamiento con prednisona a dosis de 1 mg/kg/día, con una evolución inicial favorable y la desaparición de las lesiones.
Un año después de la primera consulta el paciente refería parestesias en las extremidades inferiores, asociadas nuevamente a petequias en dicha localización, presentando en esta ocasión síndrome nefrítico. En la analítica destacaban: hipocomplementemia, criocrito del 17% y proteinograma con débil banda anómala en zona gamma. Inmunoelectroforesis sérica: componente de movilidad restringida IgM kappa (en la orina no había ningún elemento sospechoso de monoclonalidad). La función renal presentaba una creatinina de 1,2 mg/dl, sedimento con hematíes +++ y proteinuria de 2,8 g/día.
La biopsia renal confirmó la presencia de glomerulonefritis con patrón mesangiocapilar (figura 1). Se añadió al tratamiento azatioprina, con mantenimiento de la función renal y proteinurias alrededor de 1 g.
Tras seis años de seguimiento, se evidenció en un control analítico inmunofenotipo de sangre periférica compatible con LLP. Se corroboró posteriormente este hallazgo con un aspirado de médula ósea en el que se observó infiltración medular por síndrome linfoproliferativo crónico de célula pequeña de línea B, compatible con LLP quiescente, por lo que en ese momento no fue tributario de tratamiento con quimioterapia.
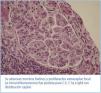
Un año más tarde el paciente presenta proteinuria persistente de 4,7 g/24 h, por lo que se decide realizar una nueva biopsia renal que confirmó la presencia de una glomerulonefritis crioglobulinémica y mostró, además, infiltración linfocitaria compatible con afectación por linfoma B de bajo grado (figura 2). Ante la afectación renal por el linfoma, se decide iniciar tratamiento con rituximab a dosis de 375 mg/m2 subcutáneo (s.c.) x 4 y tanda de recambio plasmático.
A los dos años de haber realizado tratamiento anti-CD20, el paciente se encuentra con síndrome linfoproliferativo en remisión y se mantiene la afectación renal en forma de proteinuria residual (4 g/día) con función renal conservada (Cr 0,8 mg/dl, FG: 100 ml/min) bajo tratamiento con doble bloqueo del eje renina-angiotensina-aldosterona (figura 3).
DISCUSIÓN
Además de la peculiar presentación clínica de este paciente en el que una crioglobulinemia mixta fue la primera manifestación de un LLP, este caso pone de manifiesto una rara asociación entre linfoma y glomerulonefritis por crioglobulinas, en el que destaca la ausencia de infección por VHC, como suele ser habitual en los pacientes en quienes coexisten crioglobulinemia, enfermedad glomerular y síndrome linfoproliferativo.
La mayoría de los casos de LLP presentan en suero una paraproteína IgM y en un 20% esta paraproteína puede ser una crioglobulina que resulte en fenómenos autoinmunes, como es el caso de este paciente, o una proteína que, debido la unión de las IgM con los factores de coagulación, plaquetas y fibrina, genere una coagulopatía7.
La mejor opción terapéutica en este contexto clínico consiste en la terapia antilinfoproliferativa para frenar el crecimiento de la masa tumoral y la eliminación de la paraproteína previamente secretada por el linfoma mediante recambios plasmáticos (RP).
En una serie retrospectiva multicéntrica de 33 casos de crioglobulinemia mixta en principio clasificada como idiopática se pudo establecer la causa en 20 de estos pacientes, evidenciándose después de una media de seguimiento de 67,2 meses 14 casos de enfermedad autoinmune y dos casos secundarios a infecciones no relacionadas con el VHC y cuatro pacientes con linfoma. Este estudio, al igual que nuestro caso, pone de manifiesto el valor del seguimiento a largo plazo de pacientes con crioglobulinemia idiopática, incluyendo la valoración de la función renal y el sedimento urinario8.
Otra serie descrita en la literatura mostró una serie de casos de 18 pacientes, todos con creatinina en suero elevada y un alto número presentaban proteinuria, con PBR que mostraba afectación directa del riñón por diversas neoplasias que incluían LLC/linfoma linfocítico de célula pequeña (n = 7), linfoma difuso de célula grande B (n = 6), mieloma múltiple (n = 4) y linfoma linfoblástico de célula B (n = 1).
En 10 casos (55%) coexistió una patología glomerular: cinco de ellos tenían glomerulonefritis con patrones de tipo membranopoliferativo (n = 4) y nefropatía membranosa (n=1), caracterizadas por depósito de inmunocomplejos; dos mostraron depósito de inmunoglobulina con componente monoclonal de cadenas ligeras λ amiloide (n = 1) y enfermedad por depósito de cadenas ligeras (n = 1); dos tuvieron enfermedad de cambios mínimos y en un caso hubo glomerulonefritis focal con semilunas de tipo pauciinmune. Además, una biopsia reveló nefropatía diabética y tres casos mostraron cambios isquémicos inespecíficos. En los cuatro casos restantes no hubo cambios glomerulares significativos. En 11 casos (61%), el diagnóstico de síndrome linfoproliferativo fue realizado después de la biopsia renal9.
Este caso resalta el valor de la biopsia renal como una herramienta diagnóstica para: 1) caracterizar mejor la estadificación de los linfomas con manifestaciones renales, ya que éstas pueden ser polimorfas, como en este paciente, que presentó durante el curso evolutivo desde síndrome nefrítico hasta síndrome nefrótico, y 2) como un sustrato biológico para sugerir el inicio de un tratamiento precoz en este tipo de patología hematológica.
CONCLUSIÓN
Los riñones pueden ser órgano blanco en los pacientes afectados por un LLP. La indicación de la biopsia renal cada vez más precoz en pacientes con afectación renal y LLP nos permitirá determinar otros raros patrones de daño renal que se producen en pacientes con este tipo de linfoma y evitar así el subregistro de la asociación de nefropatía y LLP.
Además, la biopsia renal en estos pacientes permitiría un diagnóstico rápido y el inicio precoz de tratamiento con quimioterapia, que es la clave de recuperación renal en pacientes con enfermedades oncohematológicas.
De lo anteriormente expuesto surge la relevancia del estudio de la función renal y del sedimento urinario en el seguimiento del paciente con linfoma, así como del trabajo en equipo con el patólogo que permite una correlación clínico-patológica de estas entidades durante todo el curso evolutivo.

Figura 1. Glomerulonefritis proliferativa con patrón mesangiocapilar.

Figura 2. A: infiltrado linfocitario denso constituido por linfocitos B compatible con linfoma de células B (hematoxilina-eosina). B: la inmunohistoquímica confirmó el diagnóstico de un linfoma linfoplasmocitario y en la figura se observa el marcaje CD20 positivo.

Figura 3. Esquema del curso clínico (seguimiento a 10 años) desde el comienzo en forma de vasculitis cutánea por crioglobulinas hasta el diagnóstico y tratamiento del linfoma linfoplasmocitario.



