Los pacientes con enfermedad renal crónica presentan una acumulación de toxinas urémicas, las cuales han sido identificadas como agentes patogénicos asociados con la mortalidad cardiovascular, muy elevada en este grupo de enfermos. Un fenómeno común a la disfunción renal progresiva y al daño vascular asociado es la acumulación anormal de proteínas de la matriz extracelular (MEC) en las estructuras renales o vasculares.
ObjetivoEstudiar la contribución de la uremia o las toxinas urémicas a la producción de citocinas y MEC en aortas de animales urémicos o células de músculo liso de aorta humana (HAOSMC).
Materiales y métodosSe utilizaron ratones con uremia inducida por una dieta rica en adenina (0,2%) durante 2, 4 o 6 semanas. Se evaluó la función renal mediante la diuresis, los niveles plasmáticos de creatinina y nitrógeno ureico plasmático, y la excreción fraccional de sodio y el daño vascular mediante histología y expresión proteica por RT-qPCR. In vitro, las HAOSMC se incubaron con toxinas urémicas: p-cresol 10-100 (μg/ml) e indoxil-sulfato 25-100 (μg/ml), solas o simultáneamente. La expresión proteica se evaluó por Western blot y microscopia confocal.
ResultadosLa administración de adenina produjo un progresivo daño renal en los ratones, un engrosamiento de la pared aórtica y un incremento de la expresión de TGF-β1 y proteínas de MEC. Las toxinas a dosis altas y combinadas también indujeron expresión de TGF-β1 y proteínas de MEC por las células HAOSMC.
ConclusionesLa uremia producida por una dieta rica en adenina o las dosis altas de toxinas urémicas indujeron el depósito anormal de proteínas de MEC en las paredes vasculares o su producción por HAOSMC. La comprensión de los mecanismos que subyacen a este proceso fisiopatológico puede resultar de utilidad en la prevención del daño cardiovascular asociado a la progresión de la enfermedad renal crónica, una dolencia, de momento, irreversible y, en ocasiones, silenciosa hasta su diagnóstico en etapas avanzadas.
Patients with chronic kidney disease present with an accumulation of uraemic toxins, which have been identified as pathogenic agents associated with cardiovascular mortality, which is very high is this patient group. A phenomenon common to the progressive renal dysfunction and associated vascular damage, is the abnormal accumulation of extracellular matrix (ECM) proteins in the renal or vascular structures.
ObjectiveTo determine the contribution of uraemia or the uraemic toxins to the production of cytokinins and ECM in aortas of uraemic animals or human aortic smooth muscle cells (HASMCs).
Materials and methodsMice were used with uraemia induced by a diet rich in adenine (0.2%) for 2, 4 or 6 weeks. Kidney function was evaluated by means of urine volume, plasma levels of creatinine, urea, fractional excretion of sodium, and vascular damage using histology, as well as protein expression using RT-qPCR. The HASMCs were incubated in vitro with uraemic toxins: p-cresol 10-100 (μg/ml) and indoxyl-sulphate25-100 (μg/ml) alone or simultaneously. The protein expression was evaluated using Western blot and confocal microscopy.
ResultsThe administration of adenine produced progressive kidney damage in the mice, thickening of the aortic wall, and increasing the expression of TGF-β1 and ECM proteins. The toxins at high doses and combined also induced the expression of TGF-β1 and ECM proteins by the HASMCs.
ConclusionsThe uraemia produced by an adenine rich diet or high doses of uraemic toxins induced the abnormal deposit of ECM proteins in the vascular wall or its production by HASMCs. The understanding of the mechanisms that underlie this pathophysiological process may be useful in the prevention of cardiovascular damage associated with the progress of chronic kidney disease, a disease, at the moment that is irreversible and occasional silent until its diagnosis in advanced stages.
La enfermedad renal crónica (ERC) constituye un problema de salud de primer orden. En España, afecta aproximadamente al 14% de la población1 y, en los casos de enfermedad avanzada, el gasto sanitario que genera es desproporcionado a su prevalencia. Además, los pacientes con disfunción renal tienen un riesgo mucho más elevado de padecer enfermedades cardiovasculares que la población general, el cual no puede ser explicado únicamente por la elevada prevalencia de factores de riesgo cardiovascular clásicos tales como la hipertensión, la hiperlipidemia, la diabetes, el tabaquismo o la hipertrofia ventricular izquierda2,3, habiéndose planteado la influencia de otros muchos factores, como el estrés oxidativo inducido por la propia uremia, el conjunto de alteraciones referentes al metabolismo fosfocálcico o la acumulación de metabolitos tóxicos capaces de dañar el árbol vascular4,5. Como consecuencia de estos factores, se producen alteraciones estructurales y funcionales cardiovasculares, como la disfunción del endotelio6, la rigidez arterial o el desarrollo de lesiones de arteriosclerosis o calcificaciones vasculares7, condicionando una isquemia tisular, el daño de determinados tejidos y la muerte. Además, un fenómeno común, que contribuye a la disfunción renal progresiva y al daño vascular de la arteriosclerosis, es la acumulación de proteínas de matriz extracelular (MEC), en mayores cantidades y de forma anormal, en las estructuras renales o vasculares, lo que condiciona, ya de por sí, importantes alteraciones en la estructura del riñón y de los vasos8.
Una característica de la ERC es su progresión, que desemboca con frecuencia en una insuficiencia renal crónica. En las fases finales, se alcanza una situación clínica conocida como uremia, en la que el medio interno se encuentra totalmente alterado. Recientemente se han identificado las toxinas urémicas unidas a proteínas como el indoxil-sulfato (IS) y el para-cresil-sulfato (p-CS), las cuales no pueden ser removidas por los métodos de diálisis convencional, como agentes patogénicos asociados con la mortalidad cardiovascular en pacientes con ERC y/o diálisis9. Entre los efectos inducidos por las toxinas urémicas que se acumulan en el plasma de estos pacientes se encuentran la inflamación vascular, la disfunción endotelial y las calcificaciones vasculares, lo cual podría explicar el mal pronóstico de los pacientes en diálisis y prediálisis10. Mediante estudios in vitro, se ha observado que el IS promueve la proliferación de las células de músculo liso vascular (VSMC)11, un proceso que, unido a su migración y elevada producción y depósito de proteínas de MEC, se relaciona con la hiperplasia de la neoíntima, anormalidades en la reparación vascular y reducción severa de la luz vascular8. Además, la migración y la proliferación de las VSMC pueden ser estimuladas por la inflamación local, lo cual puede desencadenarse por el IS y otras toxinas urémicas, como los productos de glucosilación avanzados12.
El objetivo de este trabajo fue profundizar en el estudio de la contribución de las toxinas que se acumulan en el suero de pacientes urémicos al daño vascular y, en particular, al depósito anormal de MEC en el tejido vascular. Para ello hemos utilizado un modelo animal de ERC progresiva, mediante la administración de una dieta rica en adenina, o la incubación de células de músculo liso de aorta humana (HAOSMC) con toxinas urémicas.
Material y métodosModelo animal y determinacionesSe utilizaron ratones macho de 3-4 meses de edad, de la cepa C57BL/6J (Jackson Laboratory, Bar Harbor, Maine, Estados Unidos). El grupo control fue alimentado con una dieta estándar durante todo el experimento. Para la inducción de la ERC, los ratones del grupo Adenina fueron criados como sus compañeros de camada y alimentados con una dieta que contenía un 0,2% de adenina (Sigma, St Louis, MO, Estados Unidos), durante un período de 2, 4 o 6 semanas13. Las muestras de sangre basales (semana 0) o de 2 y 4 semanas se recolectaron por incisión de una vena palpebral inferior. Todos los demás parámetros fueron analizados en las semanas 0, 2, 4 y 6 del período experimental, cuando los ratones fueron sacrificados. Un día antes del sacrificio, los animales fueron alojados en jaulas metabólicas para recoger orina de 24h. A las 24h, los ratones fueron anestesiados con ketamina-xilacina (100/20mg/kg, intraperitoneal), sometidos a una toracotomía y luego exsanguinados a través de un corte en la aurícula derecha. Se retiraron las aortas, que fueron fijadas en solución tamponada de formol al 10%, deshidratadas e incluidas en parafina de acuerdo con la técnica histológica convencional14, o fueron almacenadas a −80°C para los ensayos de RT-qPCR. Para este último, el ARN total de cada muestra fue extraído con TRIzol, transcripto a ADNc, y 10ng se amplificaron con sondas TaqMan para β-actina o mediante SYBR Green Master Mix (Life Technologies, Waltham, MA, Estados Unidos) con primers diseñados utilizando la base de datos de PubMed: TGF-β1, 5’-TTGCTTCAGCTCCACAGAGA-3’ (forward) y 5’-TGGTTGTAGAGGGCAAGGAC-3’ (reverse); fibronectina, 5’-TGAGCGCCCTAAAGATTCCA-3’ (forward) y 5’-TAGCCACCAGTCTCATGTGC-3’ (reverse); colágeno tipo I, 5’-TCCTGGCAACAAAGGAGACA-3’ (forward) y 5’-GGGCTCCTGGTTTTCCTTCT-3’ (reverse)14.
Los metabolitos plasmáticos y urinarios fueron determinados con kits comerciales, de acuerdo con las instrucciones del fabricante: creatinina y nitrógeno ureico plasmático (Arbor Assays LLC, Ann Arbor, MI, Estados Unidos). La concentración plasmática y urinaria del ion sodio (Sodium Assay Kit, Abcam, Cambridge, Reino Unido) se utilizó para calcular la excreción fraccional de sodio. Las determinaciones espectrofotométricas se realizaron con el Multimode Plate Reader Victor X4 (PerkinElmer, Waltham, MA, Estados Unidos).
Tratamiento con toxinas urémicas y determinacionesLas HAOSMC se obtuvieron por disociación enzimática de la aorta de 5 donantes de órganos, de acuerdo con las disposiciones legales españolas, con la aprobación del Comité Ético del Hospital Universitario de Getafe, como ha sido descrito previamente15, y fueron generosamente donadas por el Dr. Sánchez-Ferrer. Las células fueron mantenidas en medio de cultivo DMEM suplementado con 10% calf serum, L-glutamina, anfotericina, penicilina y estreptomicina. Para los experimentos, las HAOSMC, de pases 3 a 10, fueron cultivadas con suero humano (AB serum; BioWhittaker, Walkersville, MD, Estados Unidos) al 2,5% durante 24h e incubadas con los solutos urémicos p-cresol (pc) e IS (Sigma), utilizados a las concentraciones del rango urémico: IS 25 y 100μg/ml y pc 10 y 100μg/ml, solas o de manera simultánea a concentraciones bajas y altas (IS+pc), durante tiempos variables.
Finalizados los tratamientos se determinó el contenido proteico celular mediante Western blot convencional16 con anticuerpos primarios contra TGF-β1, colágeno i o fibronectina (Cell Signaling Technology, Danvers, MA, Estados Unidos), o TGF-β1 mediante inmunofluorescencia, con microscopio confocal Leica17.
Análisis estadísticoTodos los datos fueron analizados con el software GraphPad Prism. Los resultados se expresan como la media±SEM. Como el número de animales o de experimentos no fue nunca mayor de 10, las comparaciones se realizaron con estadística no paramétrica, aplicando el test de Kruskal-Wallis con Mann-Whitney post-test (datos no pareados) o el test de Friedman con Wilcoxon post-test (datos pareados). Un valor de p<0,05 fue considerado significativo.
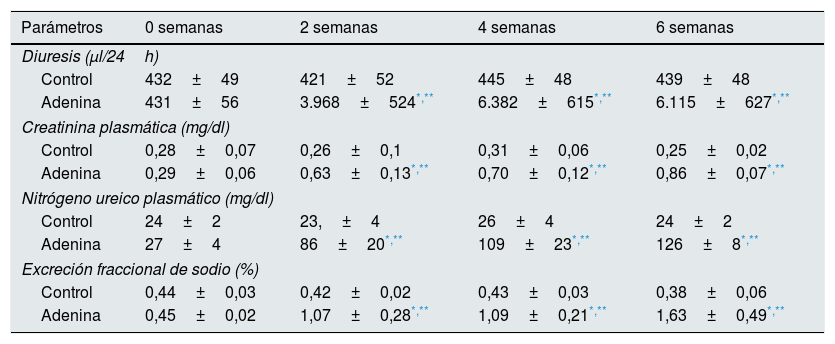
ResultadosLos ratones con dieta rica en adenina desarrollan enfermedad renal crónica y daño vascular simultáneamenteEn los experimentos realizados en ratones, se utilizó un modelo de ERC progresiva, generada por la administración de una dieta rica en adenina, durante 2, 4 o 6 semanas. En primer lugar, se comprobó el desarrollo de la ERC. Como se observa en la tabla 1, los animales alimentados con dieta rica en adenina exhibieron un progresivo aumento de las concentraciones de creatinina y nitrógeno ureico plasmáticos, y alteraciones en el manejo del sodio y el agua, manifestados por el aumento de la diuresis y la excreción fraccional de sodio. Estos cambios fueron significativos desde las 2 semanas y aumentaron progresivamente con el tiempo de administración de adenina (tabla 1). A continuación, se estudiaron las posibles alteraciones vasculares. Mediante análisis histológico se analizaron los cambios en la estructura vascular de las aortas, que se pudieran producir por la progresiva ERC. Los ratones con ERC inducida por dieta rica en adenina durante 6 semanas presentaron un engrosamiento de la media y una moderada disminución del radio de la luz (fig. 1). El análisis morfométrico indicó que el grosor de la media es significativamente mayor en los animales Adenina vs. los animales Control y que no hay diferencias significativas entre los grupos en el radio de la luz, aunque tiende a ser menor en los animales del grupo alimentado con dieta rica en adenina, en los que también se observa un aumento significativo de la ratio grosor de la media/radio de la luz. Por otra parte, los animales urémicos presentaron un incremento progresivo de la expresión de la citocina profibrótica TGF-β1, así como de las proteínas de MEC colágeno tipo i y fibronectina, marcadores de fibrosis tisular, determinados por RT-qPCR de las aortas obtenidas de los ratones (fig. 2 A-C), que fue significativo para los 3 tiempos de tratamiento en el caso del TGF-β1 y a partir de las 4 semanas para las proteínas de MEC.
Análisis comparativo de la función renal, entre los animales controles y los alimentados con adenina
| Parámetros | 0 semanas | 2 semanas | 4 semanas | 6 semanas |
|---|---|---|---|---|
| Diuresis (μl/24h) | ||||
| Control | 432±49 | 421±52 | 445±48 | 439±48 |
| Adenina | 431±56 | 3.968±524*,** | 6.382±615*,** | 6.115±627*,** |
| Creatinina plasmática (mg/dl) | ||||
| Control | 0,28±0,07 | 0,26±0,1 | 0,31±0,06 | 0,25±0,02 |
| Adenina | 0,29±0,06 | 0,63±0,13*,** | 0,70±0,12*,** | 0,86±0,07*,** |
| Nitrógeno ureico plasmático (mg/dl) | ||||
| Control | 24±2 | 23,±4 | 26±4 | 24±2 |
| Adenina | 27±4 | 86±20*,** | 109±23*,** | 126±8*,** |
| Excreción fraccional de sodio (%) | ||||
| Control | 0,44±0,03 | 0,42±0,02 | 0,43±0,03 | 0,38±0,06 |
| Adenina | 0,45±0,02 | 1,07±0,28*,** | 1,09±0,21*,** | 1,63±0,49*,** |
Los ratones fueron alimentados con dieta estándar (Control) o dieta rica en adenina (Adenina), durante 2, 4, o 6 semanas. La función renal fue determinada mediante la medición de la diuresis, la creatinina plasmática, los niveles de nitrógeno ureico plasmático y la excreción fraccional de sodio. Los valores se representan como la media±SEM.
 o con una dieta rica en adenina (Adenina). A) Las aortas fueron teñidas con hematoxilina-eosina. Se muestran cambios representativos. En los ratones Control no se aprecian lesiones. Obsérvese el engrosamiento de la media (asterisco) en las aortas de los ratones Adenina. Barra de escala: 100μm. Aumento ×20. B) Análisis morfométrico del grosor de la media y el radio de la luz, realizado mediante el software ImageJ y cálculo de su ratio. Se considera radio medio la media de 5 medidas independientes en distintas direcciones. Los valores se representan como la media±SEM. 1p<0,05 vs. Control. n=5 animales/grupo.")
Los ratones con ERC inducida por dieta rica en adenina desarrollan daño estructural vascular. Los ratones fueron alimentados durante 6 semanas con la dieta estándar (Control) o con una dieta rica en adenina (Adenina). A) Las aortas fueron teñidas con hematoxilina-eosina. Se muestran cambios representativos. En los ratones Control no se aprecian lesiones. Obsérvese el engrosamiento de la media (asterisco) en las aortas de los ratones Adenina. Barra de escala: 100μm. Aumento ×20. B) Análisis morfométrico del grosor de la media y el radio de la luz, realizado mediante el software ImageJ y cálculo de su ratio. Se considera radio medio la media de 5 medidas independientes en distintas direcciones. Los valores se representan como la media±SEM. 1p<0,05 vs. Control. n=5 animales/grupo.
 o con una dieta rica en adenina (Adenina, triángulos negros). Los niveles de ARNm de TGF-β1 (A), colágeno i (COL I, B) o fibronectina (C) en la aorta fueron determinados por RT-qPCR. Los niveles totales de β-actina se determinaron como sus respectivos controles endógenos. Los valores se representan como la media±SEM vs. Control. *p<0,05 vs. 2 semanas, 1p<0,05 vs. Control, al mismo tiempo de tratamiento. n=5 animales/grupo.")
Los ratones con ERC inducida por una dieta rica en adenina presentan un aumento de la expresión de la citocina profibrótica TGF-β1 y de genes de matriz extracelular en la aorta. Los ratones fueron alimentados durante 2, 4 o 6 semanas con la dieta estándar (Control, círculos negros) o con una dieta rica en adenina (Adenina, triángulos negros). Los niveles de ARNm de TGF-β1 (A), colágeno i (COL I, B) o fibronectina (C) en la aorta fueron determinados por RT-qPCR. Los niveles totales de β-actina se determinaron como sus respectivos controles endógenos. Los valores se representan como la media±SEM vs. Control. *p<0,05 vs. 2 semanas, 1p<0,05 vs. Control, al mismo tiempo de tratamiento. n=5 animales/grupo.
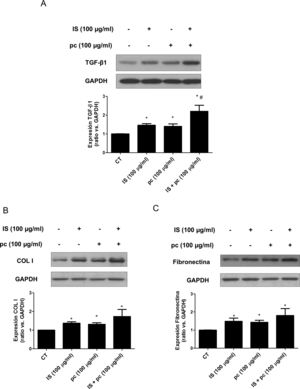
Para profundizar en los mecanismos implicados en el desarrollo de los procesos observados se realizaron ensayos in vitro, en células HAOSMC. Para ello utilizamos un modelo que tenemos puesto a punto en nuestro laboratorio, simulando en parte un entorno similar al suero urémico de los pacientes con ERC inicial o avanzada, mediante la adición al medio de cultivo de dosis bajas, altas o mezclas de 2 toxinas urémicas: el IS y el pc. En primer lugar, comprobamos que se mantuviera la viabilidad de esta línea celular frente al tratamiento con las toxinas urémicas, a las dosis y tiempos utilizados, mediante ensayos de MTT (datos no mostrados). En segundo lugar, observamos que las toxinas urémicas inducen un aumento dependiente de tiempo y dosis de la expresión de TGF-β1, producido por la incubación con las dosis altas, tanto solas como combinadas, determinado mediante Western blot e inmunofluorescencia (fig. 3 A y B), significativo a partir de las 4h de tratamiento. Este efecto es aditivo, ya que la incubación con las dosis altas de toxinas urémicas combinadas produce un aumento significativamente mayor comparado con el tratamiento con las toxinas por separado (Figura Suplementaria 1A). Por otra parte, la incubación con las toxinas a dosis bajas, tanto si se utilizan por separado como de manera simultánea, no induce cambios en la expresión de esta citocina (datos no mostrados). Paralelamente, la incubación con las mezclas altas de toxinas urémicas produjo un aumento de la expresión de las proteínas de MEC, colágeno i y fibronectina, significativo a partir de las 2 y 4h de tratamiento, respectivamente (fig. 3 C y D). Una vez más, el tratamiento es mayor cuando las toxinas se usan combinadas (Figura Suplementaria 1 B y C).
 o 24h (B) con toxinas urémicas: indoxil-sulfato (IS 100μg/ml), para-cresol (pc 100μg/ml) o una mezcla de ambas (IS+pc 100μg/ml). Se enseñan Western blots representativos de TGF-β1 (A), colágeno i (COL I, C) y fibronectina (D). Los niveles de GAPDH fueron determinados como sus respectivos controles endógenos. Las barras representan los valores del análisis densitométrico de los blots normalizados contra el control endógeno. La expresión de TGF-β1 se analizó mediante inmunofluorescencia (B). Se enseñan imágenes representativas de las fotografías obtenidas mediante microscopia confocal (×100). Los valores se representan como la media±SEM. *p<0,05 vs. Control (CT, células no tratadas).")
Las dosis altas de toxinas urémicas inducen la expresión de la citocina profibrótica TGF-β1 o de proteínas de matriz extracelular en células de músculo liso vascular. Las células de músculo liso de aorta humana fueron incubadas con medio suplementado con suero humano al 2,5%, durante 24h. Posteriormente fueron tratadas durante 2, 4, 6 o 24h (A, C y D) o 24h (B) con toxinas urémicas: indoxil-sulfato (IS 100μg/ml), para-cresol (pc 100μg/ml) o una mezcla de ambas (IS+pc 100μg/ml). Se enseñan Western blots representativos de TGF-β1 (A), colágeno i (COL I, C) y fibronectina (D). Los niveles de GAPDH fueron determinados como sus respectivos controles endógenos. Las barras representan los valores del análisis densitométrico de los blots normalizados contra el control endógeno. La expresión de TGF-β1 se analizó mediante inmunofluorescencia (B). Se enseñan imágenes representativas de las fotografías obtenidas mediante microscopia confocal (×100). Los valores se representan como la media±SEM. *p<0,05 vs. Control (CT, células no tratadas).
Los principales hallazgos de este estudio han sido que la uremia induce la expresión de la citocina profibrótica TGF-β1 y la acumulación de proteínas de MEC en las aortas de ratones en los cuales se ha inducido una uremia progresiva mediante un modelo de administración de dieta rica en adenina, y que las toxinas urémicas inducen la expresión de estas mismas proteínas en células HAOSMC.
A pesar de que se han realizado numerosos trabajos para conocer factores en la ERC que puedan producir enfermedades cardiovasculares, son escasos los que analizan la influencia de la uremia en los mecanismos fisiopatológicos celulares involucrados en el proceso que relaciona la acumulación de MEC en las paredes de los grandes vasos, que conlleva el desarrollo de un daño cardiovascular en estos enfermos. Diversos trabajos han demostrado la contribución de las toxinas urémicas acumuladas en el suero de los pacientes con ERC a la disfunción vascular, aunque los mecanismos involucrados permanecen en ocasiones oscuros. Así, el IS estimula la proliferación de las VSMC a través de la producción de especies reactivas del oxígeno11,18, y se ha observado que la administración de esta toxina promueve las calcificaciones y el engrosamiento de las paredes aórticas en ratas hipertensas19. En los pacientes con ERC las calcificaciones en las paredes vasculares aparecen particularmente en la media, en las fibras elásticas y alrededor de las VSMC20. Se ha observado que el depósito de matriz en las paredes de las arterias puede resultar de un proceso celular activo que implica la transdiferenciación de las VSMC en un tipo similar a los osteoblastos, que lleva al depósito de MEC de tipo osteogénico, un proceso que también es inducido por el IS in vitro21. Por otro lado, tanto el IS como el p-CS inducen desdiferenciación y fibrosis renal, vía transición epitelio-mesénquima, tanto in vitro como in vivo22,23. Sin embargo, hasta donde llega nuestro conocimiento, este es el primer trabajo que relaciona directamente estas toxinas urémicas con el aumento de producción de citocinas profibróticas y de MEC de tipo fibrilar por parte de las HAOSMC.
Por otro lado, en lo referente a los autacoides implicados en la progresión de la ERC, múltiples estudios han demostrado la importancia del TGF-β1 en este proceso24. En este sentido, nuestro grupo ha estado interesado desde hace años en el estudio de los mecanismos responsables de la progresión y del daño tisular en las enfermedades cardiovasculares y renales. La aproximación conceptual al estudio de estos mecanismos se ha realizado desde distintas perspectivas, pero una de las posibilidades estudiadas es que la acumulación de proteínas de MEC, a nivel renal o vascular, puede condicionar una retroalimentación positiva, que conlleva una progresión del problema o un mayor daño tisular. Hemos publicado que el contacto de las células mesangiales renales con colágenos anormales induce la síntesis de TGF-β125 y el aumento de la producción de TGF-β1 estimulado por los radicales libres17, y que en ambos casos, el proceso fibrótico se autoperpetúa, incrementando la expresión de proteínas de MEC, como el colágeno i y iv y la fibronectina17,25, lo que podría condicionar la progresión de la ERC en los pacientes. En este trabajo hemos relacionado directamente solutos que se acumulan en el suero de pacientes urémicos como son las toxinas urémicas unidas a proteínas IS y el pc, con producción de TGF-β1 y MEC por parte de las HAOSMC. Un posible mecanismo implicado en el origen de este fenómeno sería el incremento de las especies reactivas del oxígeno inducido por las toxinas urémicas, un fenómeno descrito para células endoteliales vasculares por otros y nosotros8,16, en monocitos y macrófagos8, en células de túbulo renal20 y en VSMC de ratas18, y que nosotros hemos comprobado en el modelo de uremia in vitro utilizado en este trabajo (datos no mostrados). Además, nosotros exploramos el efecto aditivo de la combinación de ambas toxinas, que presentaron un efecto sinérgico cuando se utilizaron juntas, como ha sido observado previamente para el p-CS combinado con el para-cresil glucorónido26. Una posible limitación en la traslación clínica de alguno de nuestros resultados es la utilización del pc actuando como un sustituto del p-CS, un derivado del pc por sulfatación que es su principal metabolito circulante. El pc fue durante mucho tiempo considerado una de las principales toxinas urémicas, pero actualmente se sabe que circula libre en plasma en muy baja cantidad, y que los principales cresoles que realmente son retenidos en la uremia son el p-CS y el para-cresil glucorónido, el otro conjugado del pc27. Por otro lado, hemos utilizado un modelo de uremia in vivo mediante la ingesta a largo plazo de cantidades excesivas de adenina, lo que causa nefrotoxicidad al ser metabolizada a 2,8-dihidroxiadenina, un compuesto insoluble que precipita en los túbulos renales como cristales, produciendo oclusión de los túbulos, inflamación y fibrosis tubuloinstersticial13. Dado que las alteraciones funcionales y estructurales que se desarrollan son similares a las de la ERC y ocurren de manera gradual y progresiva, este modelo nos ha resultado muy útil para examinar los mecanismos fisiopatológicos que causan enfermedad cardiovascular en la ERC. De hecho, en las aortas de los animales hemos observado un engrosamiento de la media, que también se observa en arterias coronarias de pacientes urémicos, así como el engrosamiento de la íntima-media en arterias carótidas8. Además, encontramos un progresivo acúmulo de proteínas de MEC en las paredes aórticas, un proceso que se ha relacionado, junto con la disminución en los niveles de elastina, con el remodelado arterial que se caracteriza por la rigidez de la aorta y las arterias mayores, cuya principal consecuencia fisiopatológica es la pérdida de la distensibilidad arterial8.
Finalmente, las concentraciones de toxinas que utilizamos en este estudio se encuentran dentro del rango reportado para pacientes urémicos (1-100μg/ml)28, pudiendo alcanzar valores totales en suero de 236 y 105μg/ml para el IS y p-CS, respectivamente, en pacientes en hemodiálisis28, y menores de 4,9 y 46μg/ml para el IS y p-CS, respectivamente, en pacientes con ERC que no están en diálisis, para los cuales hay muy escasa información16. Además, se han observado altos niveles de ambas toxinas en pacientes en estadios 1-5 de ERC, cuando se comparan progresadores vs. no progresadores16. Nuestros resultados, tanto in vitro como in vivo, indican que es a las dosis mayores, o a los tiempos a partir de 4 semanas de adenina, cuando se producen incrementos significativos en la producción y/o acúmulo de proteínas de MEC.
ConclusiónEn el presente trabajo se demuestra por primera vez que las toxinas urémicas que se acumulan en los pacientes con ERC en estado terminal inducen la expresión de la citocina TGF-β1 y de proteínas de MEC por parte de las HAOSMC, lo que podría explicar el mecanismo por el cual se está produciendo el daño vascular y la acumulación de MEC encontrados in vivo, en el modelo de ERC progresiva inducida por dieta rica en adenina.
FinanciaciónEl presente trabajo ha sido financiado por el Instituto de Salud Carlos III (ISCIII) y fondos FEDER (PI14/02075, PI17/00625, PI17/01513), fondos FEDER y ISCIII programas RETIC REDinREN (RD12/0021/0006 y RD16/0009/0018), Comunidad Autónoma de Madrid (NOVELREN-CM REF: B2017/BMD-3751), Universidad de Alcalá (CCG2016/BIO-043) y la Fundación Senefro (Senefro-2016) a LCB, y Fundación Renal Iñigo Álvarez de Toledo (FRIAT).
AutoríaDiego Rodríguez-Puyol y Laura Calleros comparten la dirección de este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.