




Patients with chronic kidney disease present with an accumulation of uremic toxins, which have been identified as pathogenic agents associated with cardiovascular mortality, which is very high is this patient group. A phenomenon common to the progressive renal dysfunction and associated vascular damage, is the abnormal accumulation of extracellular matrix (ECM) proteins in the renal or vascular structures.
ObjectiveTo determine the contribution of uremia or the uremic toxins to the production of cytokinins and ECM in aortas of uremic animals or human aortic smooth muscle cells (HASMCs).
Materials and methodsMice were used with uremia induced by a diet rich in adenine (0.2%) for 2, 4 or 6 weeks. Kidney function was evaluated by means of urine volume, plasma levels of creatinine, urea, fractional excretion of sodium, and vascular damage using histology, as well as protein expression using RT-qPCR. The HASMCs were incubated in vitro with uremic toxins: p-cresol 10–100 (μg/ml) and indoxyl-sulphate 25–100 (μg/ml) alone or simultaneously. The protein expression was evaluated using Western blot and confocal microscopy.
ResultsThe administration of adenine produced progressive kidney damage in the mice, thickening of the aortic wall, and increasing the expression of TGF-β1 and ECM proteins. The toxins at high doses and combined also induced the expression of TGF-β1 and ECM proteins by the HASMCs.
ConclusionsThe uremia produced by an adenine rich diet or high doses of uremic toxins induced the abnormal deposit of ECM proteins in the vascular wall or its production by HASMCs. The understanding of the mechanisms that underlie this pathophysiological process may be useful in the prevention of cardiovascular damage associated with the progress of chronic kidney disease, a disease, at the moment that is irreversible and occasional silent until its diagnosis in advanced stages.
Los pacientes con enfermedad renal crónica presentan una acumulación de toxinas urémicas, las cuales han sido identificadas como agentes patogénicos asociados con la mortalidad cardiovascular, muy elevada en este grupo de enfermos. Un fenómeno común a la disfunción renal progresiva y al daño vascular asociado es la acumulación anormal de proteínas de la matriz extracelular (MEC) en las estructuras renales o vasculares.
ObjetivoEstudiar la contribución de la uremia o las toxinas urémicas a la producción de citocinas y MEC en aortas de animales urémicos o células de músculo liso de aorta humana (HAOSMC).
Materiales y métodosSe utilizaron ratones con uremia inducida por una dieta rica en adenina (0,2%) durante 2, 4 o 6 semanas. Se evaluó la función renal mediante la diuresis, los niveles plasmáticos de creatinina y nitrógeno ureico plasmático, y la excreción fraccional de sodio y el daño vascular mediante histología y expresión proteica por RT-qPCR. In vitro, las HAOSMC se incubaron con toxinas urémicas: p-cresol 10-100 (μg/ml) e indoxil-sulfato 25-100 (μg/ml), solas o simultáneamente. La expresión proteica se evaluó por Western blot y microscopia confocal.
ResultadosLa administración de adenina produjo un progresivo daño renal en los ratones, un engrosamiento de la pared aórtica y un incremento de la expresión de TGF-β1 y proteínas de MEC. Las toxinas a dosis altas y combinadas también indujeron expresión de TGF-β1 y proteínas de MEC por las células HAOSMC.
ConclusionesLa uremia producida por una dieta rica en adenina o las dosis altas de toxinas urémicas indujeron el depósito anormal de proteínas de MEC en las paredes vasculares o su producción por HAOSMC. La comprensión de los mecanismos que subyacen a este proceso fisiopatológico puede resultar de utilidad en la prevención del daño cardiovascular asociado a la progresión de la enfermedad renal crónica, una dolencia, de momento, irreversible y, en ocasiones, silenciosa hasta su diagnóstico en etapas avanzadas.
Chronic kidney disease (CKD) is a major health problem. In Spain, it affects approximately to 14% of the population1 and, in cases of advanced disease, the health cost is disproportionate to its prevalence. It is Important to note that as compared with the general population, patients with CKD have a much higher risk of cardiovascular disease. This can not be explained solely by the high prevalence of classic cardiovascular risk factors such as hypertension, hyperlipidemia, diabetes, smoking or left ventricular hypertrophy.2,3 It has been proposed that in CKD, cardiovascular disease is influenced by many other factors such as oxidative stress, which is induced by the uremia itself, alterations of phosphate and calcium metabolism or the accumulation of toxic metabolites capable of producing vascular damage.4,5 All these factors produce structural and functional alterations of the cardiovascular system such as endothelium dysfunction,6 arterial rigidity, the development of arteriosclerotic lesions or vascular calcifications,7 conditioning tissue ischemia, tissue damage and death. In addition, a common phenomenon, which contributes to progressive renal dysfunction and the vascular damage of arteriosclerosis, is the abnormal accumulation of extracellular matrix (ECM) proteins in large quantities in the renal or vascular structures, which itself causes important alterations in the structure of the kidney and vessels.8
A characteristic of CKD is the progressive deterioration of renal function, which often leads to end stage renal disease. In the final stages of CKD, uremia occurs, a situation in which the internal environment is totally altered. Recently identified uremic toxins bound to proteins such as indoxyl sulfate (IS) and para-cresyl sulfate (p-CS) can not be removed by conventional dialysis methods and these are pathogenic agents associated with the cardiovascular mortality in CKD patients and those with more advanced kidney diseases requiring dialysis.9 Among the undesirable effects of uremic toxins, that are accumulated in the plasma of these patients, are vascular inflammation, endothelial dysfunction and vascular calcifications, which could explain the poor prognosis of dialysis and predialysis patients.10 Through in vitro studies, it has been observed that IS promotes the proliferation of vascular smooth muscle cells (VSMC),11 that together migration and high production of ECM proteins, produces hyperplasia of the neointima, abnormalities in vascular repair and marked reduction of vascular lummen.8 In addition, the migration and proliferation of VSMC can be stimulated by local inflammation, which can be triggered by IS and other uremic toxins, such as advanced glycation products.12
The aim of this work was a comprehensive study of the contribution of toxins accumulated in the serum of uremic patients to the vascular damage and, in particular, to the abnormal deposition of ECM in vascular tissue. For this purpose, we have used an animal model of progressive CKD, by feeding the animals an adenine rich diet, or by incubating human aortic smooth muscle cells (HAOSMC) with uremic toxins.
MethodsAnimal model and determinationsWe used 3–4 month old male mice strain C57BL/6J (Jackson Laboratory, Bar Harbor, Maine, United States). The control group were mice fed a standard diet during the entire experimental period. In the CKD group, mice were bred as their littermates and were fed a diet containing 0.2% adenine (Sigma, St Louis, MO, United States), for a period of 2, 4 or 6 weeks.13 Basal (week 0) or 2 and 4 week blood samples were collected by incision of a lower palpebral vein. All other parameters were analyzed at weeks 0, 2, 4 and 6 of the experimental period, when the mice were euthanized. One day before sacrifice, the animals were housed in metabolic cages to collect 24h urine. Then, the mice were anesthetized with ketamine–xylazine (100/20mg/kg, i.p.), underwent a thoracotomy and were exsanguinated through a cut in the right atrium. The aorta was removed and fixed in a buffered solution of 10% formaldehyde, dehydrated and included in paraffin according to the conventional histological technique,14 or it was stored at −80°C for the RT-qPCR assays. For the latter, the total RNA of each sample was extracted with TRIzol, transcribed into cDNA, and 10ng were amplified with TaqMan probes for β-actin or by SYBR Green Master Mix (Life Technologies, Waltham, MA, United States) with designed primers using the PubMed database: TGF-β1, 5′-TTGCTTCAGCTCCACAGAGA-3′ (forward) and 5′-TGGTTGTAGAGGGCAAGGAC-3′ (reverse); fibronectin, 5′-TGAGCGCCCTAAAGATTCCA-3′ (forward) and 5′-TAGCCACCAGTCTCATGTGC-3′ (reverse); collagen type I, 5′-TCCTGGCAACAAAGGAGACA-3′ (forward) and 5′-GGGCTCCTGGTTTTCCTTCT-3′ (reverse).14
The plasma and urinary chemistries, creatinine and the plasma urea nitrogen, were measured using commercial kits, according to the manufacturer's instructions (Arbor Assays LLC, Ann Arbor, MI, United States). The values of plasma and urinary concentration of the sodium ion (Sodium Assay Kit, Abcam, Cambridge, United Kingdom) were used to calculate the fractional excretion of sodium. The spectrophotometric determinations were carried out with the Victor Multimode Plate Reader Victor X4 (PerkinElmer, Waltham, MA, United States).
Incubation with uremic toxins. Parameters measuredThe HAOSMC were obtained by enzymatic dissociation of the aorta from 5 organ donors, in accordance with the Spanish legal provisions, with the approval of the Ethical Committee of the University Hospital of Getafe as previously described.15 These were generously donated by Dr. Sánchez-Ferrer. The cells were maintained in DMEM culture medium supplemented with 10% calf serum, l-glutamine, amphotericin, penicillin and streptomycin. For the experiments, the HAOSMC, passages 3–10, were cultured with 2.5% human serum (AB serum; BioWhittaker, Walkersville, MD, United States) for 24h and incubated with the uremic solutes p-cresol (pc) and IS (Sigma), at concentrations of the uremic range (IS: 25 and 100μg/ml and pc 10 and 100μg/ml), alone or combined together at low or high concentrations during a variable period of time.
Once the treatments were finished, the cell protein content was quantified by conventional Western blot16 using primary antibodies against TGF-β1, collagen I or fibronectin (Cell Signaling Technology, Danvers, MA, United States). The TGF-β1 was also evaluated by immunofluorescence using confocal microscopy (Leica17).
Statistic analysisAll the data was analyzed with the GraphPad Prism software. The results are expressed as the mean±SEM. As the number of animals or experiments was never greater than 10, the comparisons were made for nonparametric data, applying the Kruskal–Wallis test with Mann–Whitney post-test (unpaired data) or the Friedman test with Wilcoxon post test (paired data). A p value <0.05 was considered significant.
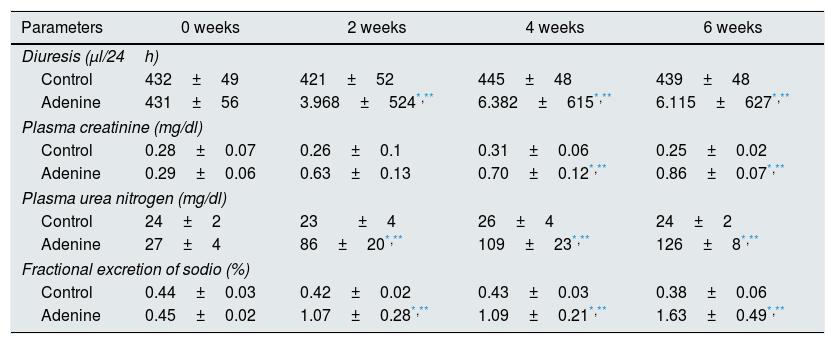
ResultsMice feed an adenine rich diet simultaneously develop chronic kidney disease and vascular damage.In the mice experiments, progressive CKD was generated by feeding the animals an adenine rich diet and the measurements were obtained at 2, 4 or 6 weeks. First, the generation of CKD was illustrated by the change in parameters that are presented in Table 1. Animals fed adenine rich diet exhibited a progressive increase in plasma creatinine and urea nitrogen concentrations, as well as alterations in the handling of sodium and water, manifested by the increase in diuresis and in the fractional excretion of sodium. These changes were significant at 2 weeks and progressively worsens with the prolongation of the adenine administration (Table 1). Next, the possible vascular abnormalities were evaluated. The changes in the vascular structure of the aortas that could be produced by progressive CKD were investigated by microscopic examination. Mice with CKD induced by 6 weeks of adenine rich diet presented a thickening of the media layer and a moderate decrease of the radius of lummen (Fig. 1). The morphometric analysis indicated that the thickness of the media is significantly higher in animals on Adenine than Controls. In these two groups, the radius of the lumen was not significantly different although it tends to be reduced in animals on adenine rich diet. Therefore, in these mice there was significant increase in the ratio media/radius of lummen. In addition, uremic animals showed a progressive increase in the expression of the profibrotic cytokine TGF-β1, as well as the ECM proteins collagen type I and fibronectin, tissue fibrosis markers as determined by RT-qPCR of the mice aortas (Fig. 2A and C), which was significant for the 3 time periods in the case of TGF-β1 and from week 4 for the ECM proteins.
Comparison of renal function in control animals vs. those on rich adenine diet.
| Parameters | 0 weeks | 2 weeks | 4 weeks | 6 weeks |
|---|---|---|---|---|
| Diuresis (μl/24h) | ||||
| Control | 432±49 | 421±52 | 445±48 | 439±48 |
| Adenine | 431±56 | 3.968±524*,** | 6.382±615*,** | 6.115±627*,** |
| Plasma creatinine (mg/dl) | ||||
| Control | 0.28±0.07 | 0.26±0.1 | 0.31±0.06 | 0.25±0.02 |
| Adenine | 0.29±0.06 | 0.63±0.13 | 0.70±0.12*,** | 0.86±0.07*,** |
| Plasma urea nitrogen (mg/dl) | ||||
| Control | 24±2 | 23±4 | 26±4 | 24±2 |
| Adenine | 27±4 | 86±20*,** | 109±23*,** | 126±8*,** |
| Fractional excretion of sodio (%) | ||||
| Control | 0.44±0.03 | 0.42±0.02 | 0.43±0.03 | 0.38±0.06 |
| Adenine | 0.45±0.02 | 1.07±0.28*,** | 1.09±0.21*,** | 1.63±0.49*,** |
The mice were fed a standard diet (Control) or a diet rich in adenine (adenine), for 2, 4, or 6 weeks. Renal function was assessed by measuring diuresis, plasma creatinine, plasma urea nitrogen levels, and fractional excretion sodium. The values are represented as the mean±SEM.
 or rich in adenine (adenine) during 6 weeks. (A) The aortas were stained with hematoxylin-eosin. Representative changes are shown. In Control mice, no lesions are seen. Note the thickening of the media (asterisk) of the aortas of the mice fed with Adenine. Scale bar: 100μm. Magnification 20×. (B) Morphometric analysis of the thickness of the media and the radius of the lumen, made by the ImageJ software and calculation of its ratio. The mean of 5 independent measurements in different directions is considered mean radius. The values are presented as the mean±SEM. 1p<0.05 vs. Control. n=5 animals/group.")
Mice with CKD induced by adenine-rich diet develop vascular structural damage. Mice were fed standard diet (Control) or rich in adenine (adenine) during 6 weeks. (A) The aortas were stained with hematoxylin-eosin. Representative changes are shown. In Control mice, no lesions are seen. Note the thickening of the media (asterisk) of the aortas of the mice fed with Adenine. Scale bar: 100μm. Magnification 20×. (B) Morphometric analysis of the thickness of the media and the radius of the lumen, made by the ImageJ software and calculation of its ratio. The mean of 5 independent measurements in different directions is considered mean radius. The values are presented as the mean±SEM. 1p<0.05 vs. Control. n=5 animals/group.
 or with a diet rich in adenine (Adenine, black triangles). The mRNA levels of TGF-β1 (A), collagen I (COL I, B) or fibronectin (C) in the aorta were determined by RT-qPCR. The total β-actin levels were as their respective endogenous controls. The values are presented as the mean±SEM vs. Control. * p<0.05 vs. 2 weeks, 1p<0.05 vs. Control, at the same time period. n=5 animals/group.")
Mice with CKD induced by a adenine rich diet show the aorta with increased expression of the profibrotic cytokine TGF-β1 and in the extracellular matrix genes. The mice were fed for 2, 4 or 6 weeks with either the standard diet (Control, black circles) or with a diet rich in adenine (Adenine, black triangles). The mRNA levels of TGF-β1 (A), collagen I (COL I, B) or fibronectin (C) in the aorta were determined by RT-qPCR. The total β-actin levels were as their respective endogenous controls. The values are presented as the mean±SEM vs. Control. * p<0.05 vs. 2 weeks, 1p<0.05 vs. Control, at the same time period. n=5 animals/group.
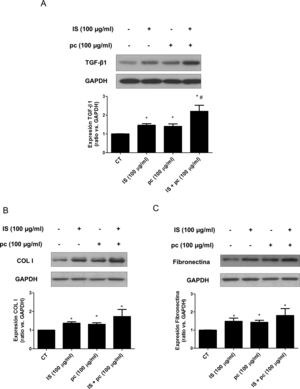
In vitro tests were performed using HAOSMC cells aiming to identify the mechanisms whereby uremic rats developed thickening of the aorta's wall. We use a model developed in our laboratory that simulate in part the environment of uremia in moderate or advanced CKD by adding low, high doses or mixtures of 2 uremic toxins (IS and pc) to the culture medium. First, cell viability assessed by MTT assays, was demonstrated while on treatment with uremic toxins, at the doses and times used, by means of (data not shown). Second, it was observed that uremic toxins induce a time and dose-dependent increase in TGF-β1 expression, after incubation with high doses, of one or the two uremic toxins combined, as determined by Western blot and immunofluorescence (Fig. 3A, B). The increase in TGF-β1 expression was significant at 4h of treatment. The effect of both uremic toxins on TGF-β1 expression is additive, since the incubation with high doses of the two compounds simultaneously produces a significantly further increase in TGF-β1 expression as compared with the separate treatment with each of the toxins (Supplementary Fig. 1A). On the other hand, the incubation with the toxins at low doses, whether used separately or simultaneously, does not induce changes in the expression of this cytokine (data not shown). In parallel, the incubation with the high concentration of uremic toxins produced an increase in the expression of the ECN proteins, collagen I and fibronectin, that is significant at 2 and 4h of treatment, respectively (Fig. 3C and D). Once again, the effect is greater if the toxins are used in combination (Supplementary Fig. 1B and C).
 or 24h (B) with uremic toxins: indoxil-sulfate (IS 100μg/ml), para-cresol (pc 100μg/ml) or a mixture of both (IS+1pc 100μg/ml). Representative Western blots of TGF-β1 (A), collagen I (COL I, C) and fibronectin (D) are shwon. The levels of GAPDH were determined as their respective endogenous controls. The bars represent the values of the densitometric analysis of the normalized blots against the endogenous control. The expression of TGF-β1 was analyzed by immunofluorescence (B). Representative images of the photographs obtained by confocal microscopy (100×) are shown. The values are represented as the mean±SEM. * p<0.05 vs. Control (CT, untreated cells).")
High doses of uremic toxins induce the expression of the profibrotic cytokine TGF-β1 and extracellular matrix proteins in vascular smooth muscle cells. Human aortic smooth muscle cells were incubated with medium supplemented with 2.5% human serum for 24h. Subsequently they were treated for 2, 4, 6 or 24h (A, C and D) or 24h (B) with uremic toxins: indoxil-sulfate (IS 100μg/ml), para-cresol (pc 100μg/ml) or a mixture of both (IS+1pc 100μg/ml). Representative Western blots of TGF-β1 (A), collagen I (COL I, C) and fibronectin (D) are shwon. The levels of GAPDH were determined as their respective endogenous controls. The bars represent the values of the densitometric analysis of the normalized blots against the endogenous control. The expression of TGF-β1 was analyzed by immunofluorescence (B). Representative images of the photographs obtained by confocal microscopy (100×) are shown. The values are represented as the mean±SEM. * p<0.05 vs. Control (CT, untreated cells).
The main findings of this study are that uremia induces the expression of the profibrotic cytokine TGF-β1 with the accumulation of ECM proteins in the aortas of mice with progressive uremia induced by adenine rich diet. Also, we show that uremic toxins induce the expression of these proteins in cultured HAOSMC cells.
Although there have been numerous studies aiming to identify CKD factors that could produce cardiovascular diseases, there are few studies analyzing pathophysiological mechanisms whereby uremia induces accumulation of ECM proteins in the wall of large vessels leading cardiovascular damage in CKD patients. Several studies have demonstrated the contribution of uremic toxins to vascular dysfunction, but often the mechanisms involved are not clear. It has been shown that IS stimulates the proliferation of VSMC through the production of reactive oxygen species,11,18 and it has been observed that the administration of this toxin promotes calcification and thickening of the aortic wall in hypertensive rats.19 In CKD patients, vascular calcifications are observed particularly in the media, also in the elastic fibers and surrounding the VSMC.20 It has been observed that the deposition of matrix in the artery wall my result from an active cellular process that involves the transdifferentiation of the VSMC into a cell type similar to the osteoblasts, which leads to the deposit of ECM of osteogenic type, a process that also it is induced by IS in vitro.21 Additionally, both IS and p-CS induce cell dedifferentiation and renal fibrosis, via epithelial–mesenchymal transition, both in vitro and in vivo.22,23 However, as far as we know, the present work is the first that directly relates these uremic toxins with the increased production of profibrotic cytokines and fibrillar type ECM by HAOSMC.
Multiple studies have shown the importance of TGF-β1 in the progression of CKD.24 Our research group has been interested for years in the study of the mechanisms responsible for the development and progression of tissue damage in cardiovascular and renal diseases. The conceptual approach to the study of these mechanisms has been carried out from different perspectives, but one of the possibilities is that the accumulation of ECM proteins, at renal or vascular level, may condition a positive feedback, which leads to a worsening of the situation or a greater tissue damage. We have reported that the contact of renal mesangial cells with abnormal collagens induces the synthesis of TGF-β125 and the production of TGF-β1 is also stimulated by free radicals,17 and that in both cases, the fibrotic process self-perpetuates, increasing the expression of ECM proteins, such as collagen I and IV and fibronectin,17,25 which favor the progression of CKD. In the resent study we have shown a direct relationship between solutes that accumulate in the serum of uremic patients such as protein bound uremic toxins, IS and pc, and the production of TGF-β1 and MEC by HAOSMC. It is possible that the origin of this phenomenon is the increase of reactive oxygen species induced by uremic toxins, a phenomenon described in vascular endothelial cells by others and ourselves,8,16 in monocytes and macrophages,8 in renal tubule cells20 and in rat VSMC,18 and that we have verified in the in vitro uremia model used in this work (data not shown).
In addition, we have explored the additive effect of the two toxins, IS and pc, as previously observed when p-CS is combined with para-cresyl glucuronide.26 A possible limitation in the clinical translation of any of our results is the use of the PC acting as a substitute for p-CS, a PC derivative by sulfation which is its main circulating metabolite. PC was for a long time considered one of the main uremic toxins, but it is currently known to circulate freely in plasma in very low amounts, and that the main cresols that are actually retained in uremia are p-CS and para-cresil glucuronide, the other conjugate of pc.27 We have used a rat model of uremia in which animals are maintained on a diet containing high amounts of Adenine which causes nephrotoxicity when metabolized to 2,8-dihydroxyadenine, an insoluble compound that precipitates in the renal tubules as crystals, producing tubule occlusion, inflammation and tubuloinsterstitial fibrosis.13 Since functional and structural alterations are similar to those observed in CKD and occur gradually and progressively, this model has been very useful to examine the pathophysiological mechanisms that cause cardiovascular disease in CKD. In fact, in the aortas of these animals we have observed a thickening of the media, which is also observed in coronary arteries of uremic patients, as well as the thickening of the intima-media of carotid arteries.8 In addition, we found a progressive accumulation of ECM proteins in the aortic wall, a process that has been related, together with the decrease in elastin levels, with the arterial remodeling characterized by stiffness of the aorta and the major arteries, whose main pathophysiological consequence is the loss of arterial distensibility.8
Finally, the concentrations of toxins that we use in this study are within the range reported in uremic patients (1–100μg/ml),28 but in uremic patients the values of these uremic toxins could be has high as 236 and 105μg/ml for the IS and p-CS respectively, in hemodialysis patients,28 and less than 4.9 and 46μg/ml for IS and p-CS respectively, in patients with CKD not on dialysis, for whom there is very scarce information.16 In addition, higher levels of both toxins have been observed in patients with CKD stages 1–5 that are progressor vs. the non progressors.16 Results from in vitro and in vivo experiments, indicate that there is a significant increase in the production and/or accumulation of ECM proteins when exposed to high concentration the uremic toxins, or after 4 weeks of high adenine diet.
ConclusionThe present work demonstrates for the first time that uremic toxins that accumulate in patients with end stage kidney diseases induce the expression of the cytokine TGF-β1 and proteins of ECM by the HAOSMC. This could explain the mechanism of vascular damage and ECM accumulation observed in vivo in the model of progressive CKD induced by adenine rich diet.
FinancingThis work has been financed by the Carlos III Health Institute (ISCIII) and FEDER funds (PI14/02075, PI17/00625, PI17/01513), FEDER funds and ISCIII RETIC REDinREN programs (RD12/0021/0006 and RD16/0009/0018), Autonomous Community of Madrid (NOVELREN-CM REF: B2017/BMD-3751), University of Alcalá (CCG2016/BIO-043) and the Senefro Foundation (Senefro-2016) to LCB, and Renal Foundation Iñigo Álvarez de Toledo (FRIAT).
AuthorshipDiego Rodríguez-Puyol and Laura Calleros share the direction of this work.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Hatem-Vaquero M, de Frutos S, Luengo A, González Abajo A, Griera M, Rodríguez-Puyol M, et al. Contribución de las toxinas urémicas a la fibrosis vascular asociada a la enfermedad renal crónica. Nefrologia. 2018;38:639–646.



