







Background: Macroscopic haematuria secondary to renal cyst rupture is a frequent complication in autosomal dominant polycystic kidney disease (ADPKD). Sickle-cell disease is an autosomal recessive haemoglobinopathy that involves a qualitative anomaly of haemoglobin due to substitution of valine for the glutamic acid in the sixth position of 3-globin gene on the short arm of chromosome 11. For the full disease to be manifested, this mutation must be present on both inherited alleles. The severity of the disease is proportional to the quantity of haemoglobin S (Hb S) in the red cells; sickle-cell trait (Hb S <50%) and homozygous sickle-cell disease (Hb S >75%). In sickle-cell disease, the abnormal Hb S loses its rheological characteristics and is responsible of the various systemic manifestations including those of the kidney, such as macroscopic haematuria secondary to papilar necrosis. Despite the generally benign nature of the sickle-cell trait, several potentially serious complications have been described. Metabolic or environmental changes such as hypoxia, acidosis, dehydration, hyperosmolality or hyperthermia may transform silent sickle-cell trait into a syndrome resembling sickle-cell disease with vaso-occlusive crisis due to an accumulation of low deformable red blood cells in the microcirculation originating haematuria from papilar necrosis. On the other hand, it has been demonstrated an earlier onset of end-stage renal disease (ESRD), in blacks with ADPKD and sickle-cell trait when compared with blacks with ADPKD without the trait. Patients and methods: We studied 2 african-american families (4 patients) which presented with both ADPKD and sickle-cell trait (Hb S <50%). The diagnosis of sickle-cell trait was confirmed by haemoglobin electrophoresis. The renal volume was measured by magnetic resonance imaging (MRI). Results: The proband subject in family 1 presented frequent haematuria episodes, associated to increase of renal volume, developed very early ESRD and was dialyzed at the age of 39 years. The other 3 patients in family 2 presented different degree of renal function. Conclusions: The presence of sickle haemoglobin should be determined in african-american and west-african patients with ADPKD because it is an important prognostic factor. Co-herence of sickle-cell trait may have influence on ADPKD evolution to ESRD and other complications, such as cystic haemorrhages. MRI can identify intracystic haemorrhage and permit renal volume measure.
Antecedentes: La hematuria macroscópica derivada de la rotura de quistes renales es una manifestación habitual en la poliquistosis renal autosómica dominante (PQRAD). La enfermedad por células falciformes es una anomalía de la hemoglobina, que se hereda con carácter autosómico recesivo, consistente en la sustitución de la valina por el ácido glutámico en la posición 6 del gen de la 3-globina en el brazo corto del cromosoma 11. La gravedad de la enfermedad es proporcional a la cantidad de hemoglobina S (Hb S) en los hematíes: los heterocigotos con hemoglobina con rasgo falciforme (Hb S <50%) y los homocigotos con enfermedad por células falciformes (Hb S >75%). La presencia de hemoglobina con rasgo falciforme (Hb AS) se acompaña de manifestaciones renales, especialmente hematuria, y la necrosis papilar es la causa más frecuente de hematuria macroscópica en los pacientes heterocigotos portadores de esta hemoglobinopatía. La asociación de estas dos enfermedades hereditarias, PQRAD y hemoglobina con rasgo falciforme, se ha comunicado raramente. Se ha sugerido que los pacientes con PQRAD y hemoglobina con rasgo falciforme podían desarrollar precozmente insuficiencia renal crónica (IRC). Recientemente, se ha comunicado que la hemoglobina con rasgo falciforme es un factor de riesgo predisponente para el desarrollo de enfermedad renal crónica en afroamericanos. Pacientes y métodos: Se estudiaron 2 familias de origen afroamericano (4 pacientes) que co-heredaron la PQRAD y la hemoglobina con rasgo falciforme (heterocigotos). El diagnóstico de hemoglobina falciforme (Hb S) se realizó por electroforesis de la hemoglobina. El volumen renal se midió mediante resonancia magnética (RM). Resultados: La paciente índice, perteneciente a una de las familias, presentó episodios de hematuria macroscópica recidivantes, asociados con incremento del volumen renal y desarrollo precoz de IRC avanzada, precisando tratamiento con diálisis a los 39 años de edad. Las 3 pacientes pertenecientes a la otra familia, de tres generaciones diferentes, presentaron distintos grados de función renal. Conclusiones: En los pacientes afroamericanos y del África subsahariana con PQRAD, debe determinarse la presencia de hemoglobina falciforme, porque ésta puede ser un importante factor pronóstico. La co-herencia de PQRAD y hemoglobina con rasgo falciforme puede influir en la evolución hacia la IRC y en el desarrollo de complicaciones, como el sangrado quístico. La imagen de RM es una herramienta de utilidad para identificar las hemorragias quísticas y para medir el volumen renal.
INTRODUCCIÓN
La poliquistosis renal es una enfermedad hereditaria, autosómica dominante, producida por mutaciones en dos genes, PKD1 (brazo corto del cromosoma 16) y PKD2 (brazo largo del cromosoma 4), que se caracteriza por la presencia de quistes renales que aumentan de forma progresiva en número y tamaño, conduciendo a la insuficiencia renal terminal entre los 50-60 años de media. En la poliquistosis renal autosómica dominante (PQRAD), la hematuria macroscópica derivada de la rotura de quistes renales es una manifestación habitual1.
La drepanocitosis o enfermedad por células falciformes es una hemoglobinopatía, que se hereda con carácter autosómico recesivo, consistente en una anomalía de la hemoglobina debido a la sustitución de la valina por el ácido glutámico en la posición 6 del gen de la 3-globina en el brazo corto del cromosoma 11. La gravedad de la enfermedad es proporcional a la cantidad de hemoglobina S (Hb S) en los hematíes: los heterocigotos con hemoglobina con rasgo falciforme (Hb S <50%) y los homocigotos con enfermedad por células falciformes (Hb S >75%). En la enfermedad por células falciformes, la Hb S anormal pierde sus características reológicas y es responsable de varias manifestaciones sistémicas, incluyendo las propiamente renales, como los infartos papilares debido a las lesiones vasculares. En la enfermedad por células falciformes, la insuficiencia renal crónica (IRC) puede ser una complicación hasta en el 4-20% de los casos. La presencia de hemoglobina con rasgo falciforme puede estar asociada también con manifestaciones renales, especialmente hematuria, y la necrosis papilar es la causa más frecuente de hematuria macroscópica en los pacientes heterocigotos portadores de hemoglobina con rasgo falciforme2-6.
La asociación de estas dos enfermedades hereditarias, PQRAD y hemoglobina con rasgo falciforme, ha sido raramente comunicada en la bibliografía. En los años noventa, se comunicó que los pacientes afroamericanos con PQRAD, que presentaban, además, la hemoglobina con rasgo falciforme, podían desarrollar más precozmente IRC terminal7,8. Desde entonces no existen referencias a familias o series con esta asociación genética, a pesar de la frecuencia relativa de ambas enfermedades hereditarias. Se presentan 4 pacientes, pertenecientes a 2 familias de origen afroamericano, con co-herencia de PQRAD y hemoglobina con rasgo falciforme (heterocigotos). Una de las pacientes, tras presentar numerosos episodios de hematuria macroscópica recidivante, como hecho peculiar, desarrolló IRC avanzada a los 39 años de edad. Las otras 3 pacientes presentaron distintos grados de función renal.
PACIENTES Y MÉTODOS
El diagnóstico de PQRAD se realizó por la historia clínica familiar y las correspondientes pruebas de imagen que incluyeron ecografía, resonancia magnética (RM) y en algunos casos también tomografía computarizada (TC). Aunque no se realizaron estudios genéticos de ADN, teniendo en cuenta las características clínicas, la forma de presentación y el tiempo del diagnóstico, la PQRAD en estas familias fue, con toda probabilidad, PKD1 (cromosoma 16). La primera familia estaba formada por dos generaciones y la segunda por tres. El diagnóstico de hemoglobina falciforme (Hb S) se realizó por electroforesis de la hemoglobina en medio ácido y alcalino. El volumen renal total se determinó mediante RM sin contraste, en secuencias potenciadas T1 y T2, y mediante técnica de segmentación manual, sumando los volúmenes de ambos riñones. En todos los pacientes que presentaron hematuria recidivante se descartó la presencia de un carcinoma medular renal. En la figura 1 y en la figura 2 se representan los árboles genealógicos de ambas familias. En la figura 3, en la figura 4 y en la figura 5 se exponen imágenes representativas de los riñones poliquísticos. En la tablas 1 y en la tabla 2 se resumen las características clínicas y los datos evolutivos de los pacientes.
Caso clínico índice (caso 1)
Mujer nacida en el año 1968, afroamericana (natural de Santo Domingo) quien, a la edad de 35 años, tras presentar un episodio de cólico nefrítico con expulsión de varios coágulos, al realizarle una ecografía renal fue diagnosticada de PQRAD. En los antecedentes familiares refería que su padre, diagnosticado de PQRAD, estaba en tratamiento con hemodiálisis desde los 55 años de edad. Una hermana menor también había sido diagnosticada de PQRAD, auqnue se desconocía el grado de función renal. La madre, la hermana menor y la propia paciente eran portadoras del rasgo de hemoglobina falciforme (Hb AS). En abril de 2005, cuando se encontraba estudiando en Alemania, comenzó con dolor en fosa renal derecha, hematuria oscura con coágulos, que requirió ingreso hospitalario, siendo diagnosticada de quiste renal complicado. En ese momento tenía Crs 2 mg/dl y Hb 5,2 g/dl, por lo que precisó la transfusión de 4 concentrados de hematíes. Una semana después reingresó por dolor recidivante en la fosa renal derecha, precisando analgesia potente. Tras la realización de cistoscopia le objetivaron una masa vesical compatible con coágulos, y requirió 2 transfusiones más. Recibió tratamiento antibiótico y sintomático, presentando mejoría de la anemia hasta Hb 13,6 g/dl y la Crs 1,9 mg/dl. En octubre 2006, en una analítica de control presentaba Crs 2,4 mg/dl y aclaramiento de Cr de 29 ml/min. En la RM el volumen de los riñones fue RD = 1.392 ml y RI = 1.485 ml (volumen renal total = 2.877 ml), con varios quistes con signos de sangrado intraquístico. La electroforesis de la hemoglobina demostró la siguiente distribución: Hb S 40%, Hb A 57%, Hb A2 3% (tabla 1). Entre los años 2006 y 2008 presentó varios episodios de hematuria recidivante con coágulos acompañados de anemización, que precisaron tratamiento con múltiples transfusiones. En junio 2008 presentaba en la analítica: Crs 4,4 mg/dl y aclaramiento de Crs de 15 ml/min. En la RM el volumen de los riñones fue RD = 2.748 ml y RI = 2.076 ml (volumen renal total = 4.824 ml). Tras presentar repetidos episodios de hematuria, unos espontáneos y otro tras una caída accidental, con anemización, que no respondieron al tratamiento médico, incluyendo ácido tranexámico, se planteó una embolización que no fue aceptada por la paciente, y en septiembre de 2008 se realizó nefrectomía izquierda. A continuación precisó tratamiento con hemodiálisis a través de un catéter yugular permanente. Los intentos (en dos ocasiones) de realizar una fístula arteriovenosa interna para hemodiálisis resultaron infructuosos, al producirse la trombosis de la misma. Después de 2 años en hemodiálisis, por presentar de nuevo hematurias incoercibles, en septiembre de 2010, tuvieron que practicarse embolización y nefrectomía derecha. En ninguna de las dos piezas de nefrectomía se observaron cambios compatibles con carcinoma medular renal.
DISCUSIÓN
En la PQRAD la hematuria macroscópica derivada de la rotura de quistes renales es una manifestación habitual. Así, la hematuria es el signo de presentación en el 35% de los casos, y en el 50% de los pacientes se observa hematuria macroscópica o microscópica1. El riesgo de hematuria parece asociarse con la presencia de hipertensión arterial y con el aumento del tamaño de los quistes. Aunque la mayoría de los pacientes refieren posibles causas precipitantes como traumastismos y ejercicios violentos, no se ha demostrado de forma inequívoca una asociación precisa. Actualmente, con la extensión del uso de las técnicas de imagen, y en concreto con la RM, pueden observarse hemorragias intraquísticas que en muchos casos habían pasado totalmente desapercibidas. Estos hechos tienen gran importancia, ya que se conoce que los pacientes con PQRAD que presentan frecuentes episodios de hematuria o evidencias de hemorragias intraquísticas tienen una evolución más rápida hacia la IRC. Por otra parte, la presencia de hemoglobina con rasgo falciforme (Hb AS) se acompaña de manifestaciones renales, especialmente hematuria, siendo la necrosis papilar la causa más frecuente de hematuria macroscópica en los pacientes heterocigotos portadores de esta hemoglobinopatía9-12. No es de extrañar, por lo tanto, que la co-herencia de ambas enfermedades genéticas origine una sinergia en cuanto a la aparición de estas complicaciones hemorrágicas y en la evolución a la IRC.
En la familia 1, una de las enfermedades de herencia autosómica dominante, la PQRAD, se transmitió por línea paterna y la otra recesiva, hemoglobina con el rasgo falciforme, lo hizo por línea materna (figura 1). En esta familia, el caso índice (caso 1) fue una mujer con ambas enfermedades genéticas, que desarrolló una IRC de evolución rápida y que tuvo que comenzar tratamiento con hemodiálisis a los 39 años de edad. En esta paciente la formación y el desarrollo de los quistes renales fue muy precoz y, muy probablemente, la asociación del rasgo falciforme (Hb AS) favoreció los episodios de hematuria macroscópica recidivantes, las hemorragias intraquísticas y el desarrollo temprano de IRC avanzada. Es de destacar, en este caso, que los episodios de hematuria, en ocasiones, se vieron precedidos de un viaje en avión de varias horas de duración (lógicamente en condiciones de hipoxia relativa) o por mínimos traumatismos. Por otra parte, las imágenes secuenciales de RM con la medición de los volúmenes renales pusieron de manifiesto un extraordinario incremento de los mismos en un tiempo relativamente corto (un 68% en 2 años). Este hecho, sin duda, se debió al sangrado intraquístico y a los hematomas intrarrenales que fueron detectados en las fases más avanzadas de su enfermedad, se confirmaron con la TC y finalmente en la anatomía patológica. Esta evolución contrastó con la de su padre, que no era portador de la hemoglobina con rasgo falciforme y precisó tratamiento con hemodiálisis a partir de los 55 años. En la familia 2, ambas enfermedades, la PQRAD y la hemoglobina con rasgo falciforme, se heredaron por la misma línea (materna) (figura 2). En esta familia, la PQRAD se diagnosticó en uno de los individuos precozmente en la infancia, a los 11 años de edad (caso 4). Esta paciente y su madre (caso 3) presentaron hiperfiltración glomerular. La abuela (caso 2), que tuvo algunos episodios de hematuria macroscópica, desarrolló IRC, presentando en la RM imágenes de sangrado intraquístico y un volumen renal total moderadamente elevado. Hasta donde conocemos, éste es el primer estudio que ha evaluado a familias con esta asociación genética en Europa.
Resulta sorprendente que, hasta ahora, sólo existan en la bibliografía médica dos comunicaciones previas, las dos pertenecientes al mismo grupo, en las que se describe la asociación de ambas enfermedades genéticas, PQRAD y hemoglobina con rasgo falciforme en afroamericanos7,8. En una de ellas, los pacientes afroamericanos con PQRAD y hemoglobina con rasgo falciforme (Hb AS) mostraron una progresión más rápida hacia la IRC terminal que aquellos individuos sin el rasgo falciforme7. Por otra parte, existen muy pocos datos en la bibliografía que hayan examinado la relación entre hemoglobina con rasgo falciforme y enfermedad renal crónica. Sólo recientemente, en un estudio realizado en EE.UU., se ha comunicado una prevalencia un 50% más alta de hemoglobina con rasgo falciforme entre una población de afroamericanos con IRC terminal, comparada con la inferida del programa de detección de hemoglobinopatías en el recién nacido, hecho que sugiere que puede representar un factor de riesgo para el desarrollo de enfermedad renal crónica13,14.
El mecanismo por el que la hemoglobina con el rasgo falciforme contribuiría a la progresión de la enfermedad renal crónica en la PQRAD puede ser multifactorial15. La aparición repetida de áreas localizadas de falciformación y oclusión venosa, dando lugar a daño microvascular crónico, podría producir hipoxia crónica y fibrosis túbulo-intersticial. Es posible que la hemoglobina con rasgo falciforme, coexistiendo con otras situaciones que afectan a la microvasculatura renal, como la PQRAD, pudiera actuar sinérgicamente para acelerar el daño renal. En este sentido, hay que tener en cuenta que los niveles séricos de factores angiogénicos indicarían un estado proangiogénico en los adultos con enfermedad de células falciformes16. Cabe la posibilidad de que la producción de factores proangiogénicos estimulantes, presentes en ambas enfermedades y actuando de forma sinérgica, fueran responsables de las hemorragias quísticas y de la rápida evolución hacia la IRC en algunos casos de PQRAD.
La presencia de hemoglobina con rasgo falciforme (Hb AS) puede afectar también al curso y al cuidado de los pacientes con IRC terminal, puesto que puede ser un factor de riesgo independiente de tromboembolismo venoso entre los afroamericanos13. Esta predisposición a la trombosis podría afectar al fallo de la fístula arteriovenosa y a la pérdida de accesos vasculares, como ocurrió en nuestro caso índice.
En conclusión, en los pacientes afroamericanos y en los procedentes del África subsahariana con PQRAD debe determinarse la existencia de hemoglobina falciforme, ya que su presencia puede ser un importante factor pronóstico. La co-herencia de hemoglobina con rasgo falciforme puede influir en las complicaciones de los pacientes con PQRAD y en la evolución a la IRC. Probablemente lo mismo podría ser aplicable a otras patologías renales de gran prevalencia como la hipertensión arterial y la diabetes mellitus.
AGRADECIMIENTOS
Este estudio se llevó a cabo, en parte, con una ayuda del Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación (EC08/00236) y al Programa Intensificación Actividad Investigadora (Agencia Laín-Entralgo/CM) a R.P.

Figura 1. Árbol genealógico de la familia 1.

Figura 2. Árbol genealógico de la familia 2.

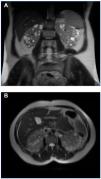
Figura 3. RM del caso 3 en la que se observan riñones que conservan su forma, con múltiples quistes pequeños (VRT = 603 ml). A) Corte coronal. B) Corte axial.

Figura 4. RM del caso 2 en la que se observan riñones aumentados de tamaño, con múltiples quistes grandes, con varios de ellos con signos de hemorragia intraquística (VRT = 1.322 ml). A) Corte coronal. B) Corte axial.

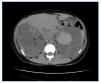
Figura 5. Corte axial de TC del caso 1 en el que se observan riñones muy aumentados de tamaño, con múltiples quistes con signos intraquísticos de hemorragia-hematoma (VRT = 2.877 ml).

Tabla 1. Características clínico-hematológicas de las 4 pacientes con PQRAD y HbAS

Tabla 2. Datos evolutivos renales de las 4 pacientes con PQRAD y HbAS



