




La activación del sistema del complemento interviene en el desarrollo de varias enfermedades renales, como las glomerulonefritis mediadas por anticuerpos, el daño por isquemia-reperfusión en los trasplantes renales o el rechazo de los injertos. Además, alteraciones en la vía alternativa están directamente implicadas en la patogénesis de las glomerulopatías C3 y del síndrome hemolítico urémico atípico. Estas alteraciones pueden ser congénitas o adquiridas; estas últimas en forma de autoanticuerpos dirigidos contra los diversos componentes y reguladores de la vía alternativa del complemento. Las consecuencias funcionales de algunos de estos anticuerpos y su asociación con estas enfermedades se conocen desde hace tiempo, pero de otros solo existen descripciones de casos aislados. En este artículo, se describen los autoanticuerpos frente a proteínas de la vía alternativa del complemento, sus características y su implicación en la enfermedad renal.
Complement system activation plays an important role in several renal pathologies, including antibody-mediated glomerulonephritis, ischaemia-reperfusion injury of trasplanted kidneys or renal allograft rejection. Besides these conditions, alternative pathway abnormalities are directly involved in the pathogenesis of C3 glomerulopathies and atypical haemolytic uraemic syndrome. These abnormalities may be inherited or acquired, the latter as autoantibodies directed against the various components and regulators of the alternative complement pathway. The functional consequences of some of these antibodies and their association with these conditions are well known, whereas for other antibodies only isolated cases have been reported. This article describes the autoantibodies that target the alternative complement pathway proteins, their characteristics and their clinical relevance in renal pathologies.
El sistema del complemento está integrado por un conjunto de más de 50 proteínas solubles y de membrana que se activan de forma secuencial en forma de cascada enzimática que debe ser estrechamente regulada. Entre las funciones más conocidas del sistema del complemento están la opsonización y eliminación de patógenos, así como la retirada de restos apoptóticos y complejos inmunes de la circulación. Además, actualmente se le reconoce un papel importante en la modulación de la respuesta inmune adaptativa y en gran número de procesos homeostáticos no relacionados con la defensa frente a infecciones. El complemento también está implicado en una amplia serie de enfermedades, bien por exceso, bien por falta de activación, bien por una regulación incorrecta1.
El sistema del complemento puede activarse por 3 vías: la clásica, la de las lectinas y la alternativa (fig. 1).
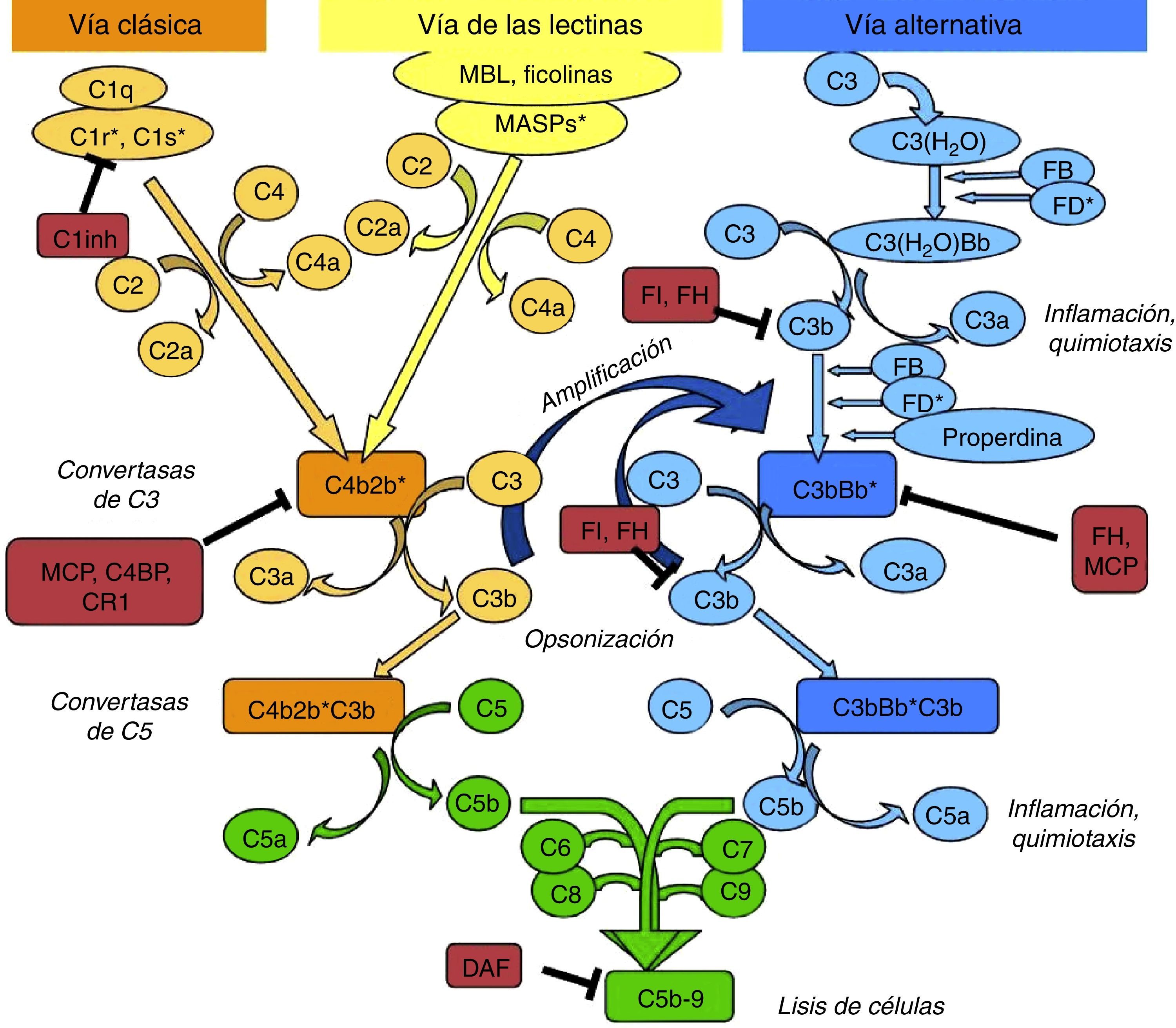
Esquema de la activación del sistema del complemento. El sistema del complemento puede ser activado por 3 vías. La activación por cualquiera de ellas lleva a la generación de una convertasa de C3 (C4b2b* o C3bBb*) que corta C3 en C3b y C3a. El C3b generado por cualquier convertasa puede formar, a su vez, más convertasa de la vía alternativa, a través de la cual se produce la amplificación de la activación del complemento. La unión de una nueva molécula de C3b a las convertasas de C3 les confiere la capacidad de cortar C5 en C5a y C5b. Este último inicia la vía terminal del complemento, que eventualmente lleva a la formación del complejo de ataque a la membrana (C5b-9), lisando las células diana. La activación del complemento está controlada a varios niveles mediante diversas proteínas reguladoras, tanto solubles como de membrana.
La vía clásica se activa fundamentalmente por la unión del C1q a complejos antígeno-anticuerpo y la de las lectinas, por la unión de MBL o ficolinas a ciertos residuos azucarados de la superficie de las bacterias. La activación de estas 2 vías da lugar a la generación de un complejo proteico con actividad enzimática: la convertasa de C3 de la vía clásica/de las lectinas (C4b2b) capaz de cortar C3 en C3a y C3b.
La vía alternativa se activa de forma continua y espontánea al hidrolizarse un enlace tioester presente en la molécula de C3. Este C3(H2O) es capaz de unirse al factor B (FB) que, una vez activado, forma la convertasa inicial de la vía alternativa que corta C3, generando C3a y C3b. Este C3b se deposita en las superficies celulares y es capaz de formar nuevas convertasas al unirse al FB (C3bBb). Esto favorece el procesamiento de más moléculas de C3 y la subsiguiente generación de más C3b, amplificando la cascada del complemento, independientemente de la vía inicial de activación. La properdina se puede unir a estos complejos, y estabilizarlos, o bien unirse directamente a la superficie de ciertos patógenos y así servir como plataforma para la formación de nuevas convertasas. Cuando a las convertasas de C3 se les une una nueva molécula de C3b, forman las convertasas de C5, capaces de cortar C5 generando C5a, una potente anafilotoxina, y C5b que sirve como iniciador para la formación del complejo de ataque a la membrana al añadirse los componentes de C6 a C9.
Para limitar la actividad de las convertasas en la superficie de las células, existen proteínas reguladoras solubles (factor H [FH], factor I [FI]) y de membrana (MCP, DAF, CR1), que diferencian a las células propias de los patógenos y las protegen del daño mediado por el complemento. En concreto, el glomérulo renal es muy vulnerable al daño inflamatorio mediado por el sistema del complemento, que puede ser debido a la presencia de fragmentos de activación circulantes (activación sistémica) o producidos in situ en el glomérulo (activación local)2. Aunque distintas enfermedades pueden estar asociadas a la activación por diferentes vías, un mecanismo patogénico común es el daño producido por la activación del C52.
La activación del complemento es de particular importancia en la patogénesis del síndrome hemolítico urémico atípico (SHUa) y en la glomerulopatía C3 (C3G). En ambos casos se encuentran mutaciones en genes de la vía alternativa, que provocan una regulación incorrecta de la activación de esta vía. Además de mutaciones, en algunos casos se encuentran autoanticuerpos dirigidos frente a componentes de la vía alternativa3, que pueden alterar el funcionamiento de sus proteínas diana, e interferir en la regulación del complemento.
A continuación, aparecen detallados los autoanticuerpos frente a proteínas de la vía alternativa descritos hasta ahora, sus principales características y sus principales repercusiones clínicas (tabla 1).
Anticuerpos frente a proteínas de la vía alternativa en enfermedades renales
| Anticuerpo | Epítopos | Efecto | Enfermedad (frecuencia) | Referencia |
|---|---|---|---|---|
| C3NeF | Convertasa de C3 (C3bBb). C3b y Bb | Estabilización de la convertasa. Consumo masivo de C3 | EDD (80%), C3G (50%), lipodistrofia adquirida, LES. Individuos sanos | 4–6,10–13 |
| Anti-FH | SCRs 18-20 | Impiden la regulación en las superficies celulares | SHUa (10%). Asociado a deficiencia de FHR-1 | 16,19,21–23,25,26 |
| SCRs 1-5 | Interfieren con la actividad cofactora de FI | C3G | 28–33 | |
| Anti-FB/(anti-C3) | FB y Bb | Estabilización de la convertasa de C3 pero inhibe la activación de la vía terminal. Consumo de C3 | EDD (un caso) | 14 |
| C3b y FB | Activación de la convertasa | EDD (2 casos) | 15 | |
| Anti-FI | FI | Sin efecto | SHUa (<2%) | 34 |
| Anti-properdina/anti-C3/anti-FB/anti-FI | Properdina, C3, FB, FI | Activación de la vía alternativa en fase fluida | Nefritis lúpica (un caso) | 35 |
C3G: glomerulopatía C3; C3NeF: factor nefrítico; EDD: enfermedad por depósitos densos; FB: factor B; FH: factor H; FI: factor I; LES: lupus eritematoso sistémico; SHUa: síndrome hemolítico urémico atípico.
El factor nefrítico (C3NeF) es un anticuerpo que se une a la convertasa de C3 de la vía alternativa4, que impide su disociación espontánea y la mantiene estable y activa durante más tiempo, incluso imposibilitando la acción de proteínas reguladoras sobre ella5 (fig. 2). Esta estabilización tiene generalmente como consecuencia un consumo masivo de C3 a nivel sistémico y una activación de la vía terminal6. Inicialmente, se consideró que estos autoanticuerpos reconocían un neoepítopo que se generaba al ensamblarse el complejo enzimático que constituye la convertasa, aunque posteriormente se ha encontrado que algunos de ellos, además, son capaces de reconocer alguno de los componentes de la convertasa de forma aislada (C3b, Bb)7.
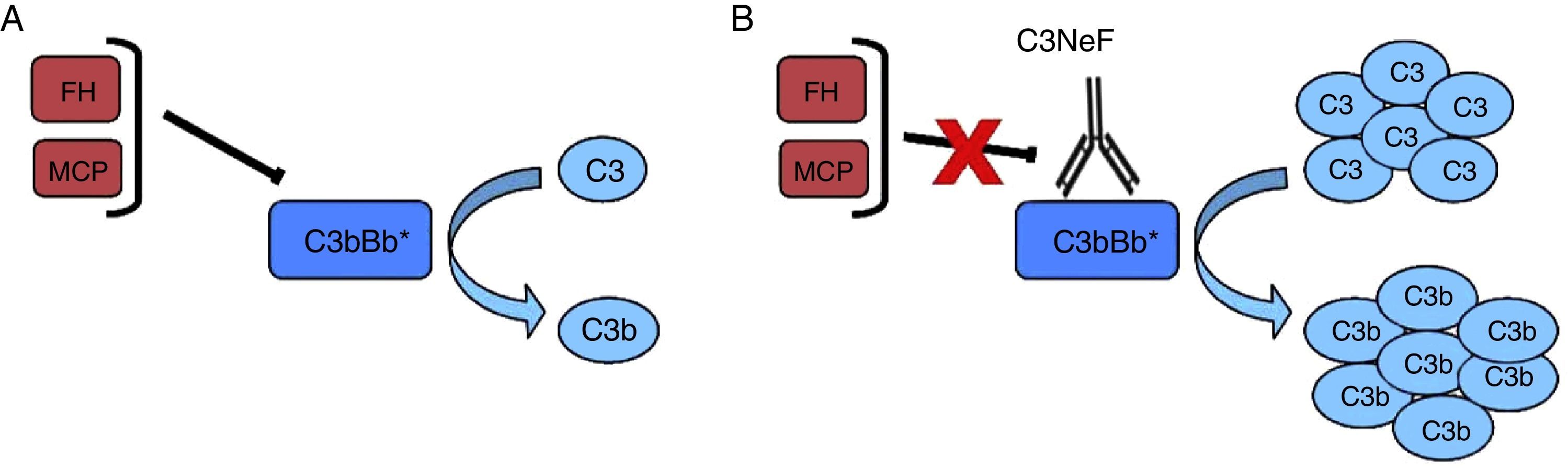
C3NeF. A) En condiciones normales, la convertasa de C3 es capaz de cortar C3 en C3b y C3a, pero existen proteínas reguladoras (FH, MCP) que favorecen su disociación y regulan su activación espontánea. B) La existencia del C3NeF estabiliza la convertasa, impide la acción de estos reguladores y permite que permanezca activa durante más tiempo y sea capaz de cortar más C3.
Los C3NeF son un grupo heterogéneo de anticuerpos, IgG o IgM, que reconocen diferentes epítopos y que producen distintos efectos sobre la activación del complemento5,6,8. Debido a esta heterogeneidad, su detección y la determinación de sus efectos sobre la regulación del complemento a veces resulta problemática.
El C3NeF está fuertemente asociado a la enfermedad por depósitos densos (EDD), que se encuentra en aproximadamente el 80% de los pacientes con esta dolencia, aunque también se encuentra presente en un 50% de los pacientes con glomerulonefritis C3 y MPGN I y III8,9, y en lipodistrofia adquirida parcial10, algunos casos de lupus eritematoso sistémico11 y de glomerulonefritis postestreptocócicas12. A pesar de esta frecuente asociación con EDD, aún no está claro si el anticuerpo es el origen de la enfermedad o si aparece de forma secundaria por la presencia continua de estos neoepítopos, ya que también se encuentra en individuos sanos13 y no se ha llegado a establecer una correlación con la clínica en los pacientes9. En cualquier caso, la activación continua de la convertasa de C3, y en algunos casos de la de C5, debido a la presencia de este anticuerpo, contribuye a la progresión de esta enfermedad.
Anticuerpos anti-C3b y anti-FBLa presencia de anticuerpos que reconocen de forma aislada alguno de los componentes de la convertasa también ha sido descrita. Inicialmente se encontró un anticuerpo anti-FB en una paciente de EDD, con C3NeF negativo por el ensayo hemolítico tradicional. Este anticuerpo reconocía el FB del suero y el fragmento Bb cuando formaba parte de la convertasa de C3, impidiendo tanto su disociación espontánea como la mediada por el FH. De esta forma inducía el consumo de C3, pero, en cambio, al contrario que la mayoría de los C3NeFs, inhibía la formación de la convertasa de C514.
Posteriormente, en 2011, se describió la presencia simultánea de anticuerpos anti-FB y anti-C3b en 2 pacientes con EDD15. Estos anticuerpos producían un aumento de la actividad de la convertasa y, por tanto, de la generación de fragmentos de activación del complemento. Al igual que en el caso de los C3NeF, no está claro aún si estos autoanticuerpos son la causa de la enfermedad o si aparecen de forma secundaria por el aumento de productos de activación que circulan en el plasma.
Anticuerpos anti-FHLos anticuerpos anti-FH asociados al SHUa se conocen desde 200516, y se estima que están presentes en aproximadamente el 10% de los casos en series de población europea17 y hasta en un 56% en una gran serie de pacientes infantiles de la India18. La presencia de autoanticuerpos anti-FH en SHUa está frecuentemente asociada a la deleción del gen de CFHR119–21. Inicialmente se determinó el epítopo que reconocen estos anticuerpos en la región C-terminal de la molécula de FH, donde se encuentran los dominios de reconocimiento y unión a superficies22,23 (fig. 3). Es en estos dominios, en los SCRs 19-20, donde además se acumulan la gran mayoría de las mutaciones en el gen de FH asociadas a SHUa24. Más recientemente se ha comprobado que, sobre todo en la fase aguda, estos anticuerpos reconocen otras regiones a lo largo de toda la molécula, formando complejos antígeno-anticuerpo y bloqueando no solo la unión de FH a las superficies celulares, sino también su actividad como cofactor del FI25.
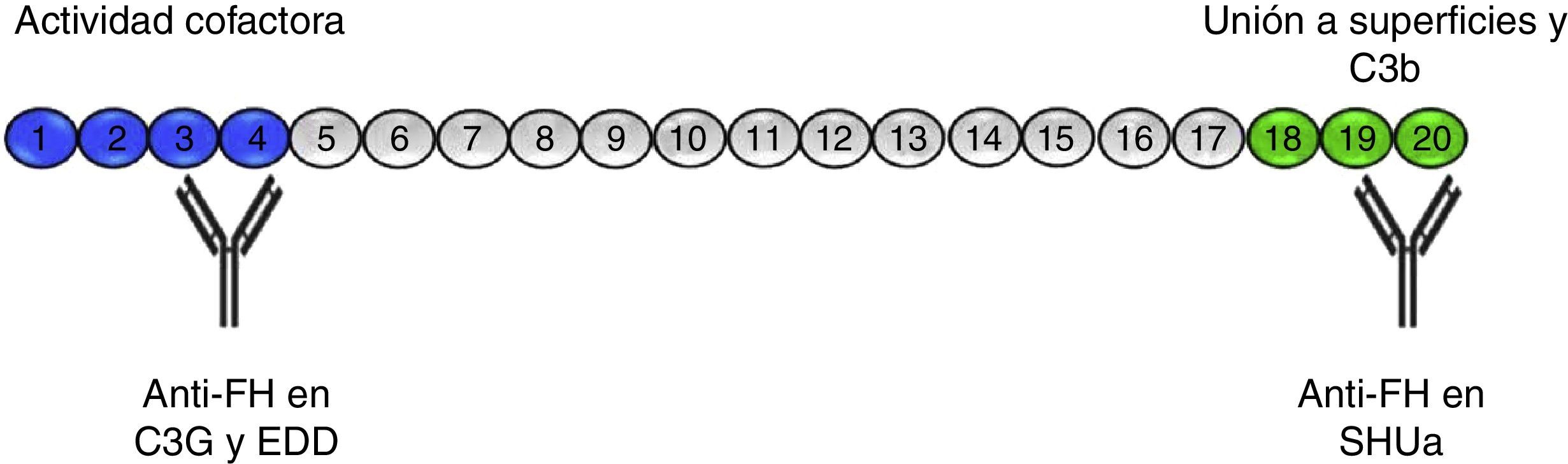
Autoanticuerpos anti-FH. El FH está formado por 20 dominios SCR. En el extremo N-terminal de la proteína (SCRs 1-4) es donde reside la actividad cofactora del FI y frente a donde están dirigidos los autoanticuerpos descritos en pacientes con C3G y EDD. A través de los SCRs 18-20 el FH se une a las superficies celulares y al C3b que se ha depositado en ellas. Los autoanticuerpos anti-FH en el SHUa reconocen esta región e impiden la unión del FH a las células endoteliales y la regulación de la vía alternativa sobre ellas.
En un trabajo reciente (2015)26, mediante el uso de fragmentos recombinantes con mutaciones puntuales en los SCRs 19-20 del FH, se ha delimitado con más precisión el epítopo que reconocen estos anticuerpos. En los pacientes con deficiencia de FHR-1 se ha encontrado que los anticuerpos anti-FH se unen a una región que adquiere una configuración diferente en FH y en el FHR-1 tras la unión a ciertos ligandos, incluyendo diversas proteínas bacterianas, lo que ha permitido proponer un modelo que explica el papel que tiene la ausencia del FHR-1 sobre el mantenimiento de la tolerancia al FH.
De una cohorte de alrededor de 400 pacientes diagnosticados de SHUa, recogidos desde el año 1999, se han seleccionado 14 de ellos por presentar anticuerpos anti-FH para estudiar las características de estos autoanticuerpos. Excepto uno, todos los demás pacientes empezaron en la infancia, lo que corresponde aproximadamente al 10% de los casos pediátricos de esta cohorte, de acuerdo con otras series.
En este grupo de pacientes se ha caracterizado la subclase y la cadena ligera de la IgG responsable de esta actividad y se ha mapeado la región del FH que es reconocida por los anticuerpos usando fragmentos de FH recombinantes que incluyen las zonas funcionalmente relevantes de la proteína. Los anticuerpos anti-FH de los pacientes de SHUa reconocen fundamentalmente la región C-terminal, especialmente los de pacientes con deficiencia de FHR-1. En el caso de los pacientes sin esta deficiencia, sus autoanticuerpos, además de reconocer esta región, se unen a otras zonas a lo largo de la proteína. En experimentos realizados con los fragmentos recombinantes que contienen las mutaciones, hemos obtenido resultados concordantes con los del grupo de Bhattacharjee et al.26, lo que apoya el modelo propuesto en la generación de estos autoanticuerpos, al menos en los pacientes con deficiencia de FHR-1. Además, se han analizado muestras seriadas obtenidas durante el seguimiento de los pacientes para comprobar si se producía algún cambio en los epítopos o en la avidez de estos anticuerpos, evidenciándose que, aunque existe heterogeneidad entre los pacientes, las características de sus autoanticuerpos permanecen constantes a lo largo del tiempo, aunque el título total suela disminuir con el tiempo. Todos estos resultados muestran una respuesta oligoclonal y restringida en la generación de estos autoanticuerpos (Nozal et al.27).
La presencia de anticuerpos anti-FH en pacientes con C3G es mucho menos frecuente que en el caso del SHUa, a pesar de estar descritos por primera vez en el año 199228. Este primer caso descrito era un «minianticuerpo» formado por dímeros de cadenas lambda monoclonales, que producía un consumo de C3 al inhibir la función reguladora del FH. Posteriormente, se comprobó que este anticuerpo reconocía el SCR3, inhibiendo su capacidad para unirse al C3b29 (fig. 3). No ha sido hasta los últimos 4 años cuando se han descrito más pacientes, aunque siguen siendo muy escasos30–33. En 3 de estos casos se ha identificado la región del FH que reconocen los anticuerpos, situándola en el dominio N-terminal, responsable de la actividad reguladora, y se ha comprobado que la fracción IgG provocaba una desregulación de la vía alternativa31,33 y, en uno de ellos, que esta desregulación era debida a que bloqueaban específicamente la actividad cofactora del FH sobre el FI31.
En este mismo año se ha publicado una serie con 17 pacientes con C3G y autoanticuerpos anti-FH34. Estos anticuerpos estaban dirigidos frente a la región N-terminal del FH, y aproximadamente un tercio de ellos alteraban la regulación en fase fluida disminuyendo la actividad cofactora, pero no la regulación en superficies, como sí ocurre en el SHUa.
En estas enfermedades no se ha encontrado asociación de los autoanticuerpos con la deleción de CFHR133, aunque sí es frecuente que se detecten simultáneamente con el C3NeF, lo que dificulta, en parte, atribuirles claramente un papel patogénico.
Aunque la presencia de estos anticuerpos en pacientes con glomerulonefritis sea mucho menos frecuente que en el SHUa, no deben descartarse como causa de desregulación del complemento, ya que, en todos los casos donde se han estudiado sus repercusiones funcionales, han resultado ser responsables de inhibir la función reguladora del FH.
Anticuerpos anti-FIEn un estudio de una cohorte de 175 pacientes con SHUa, se encontraron anticuerpos anti-FI en 3 de ellos35. La unión al FI resultó ser específica en los 3 y se encontraron también complejos circulantes en el suero de estos pacientes, pero al llevar a cabo ensayos funcionales con estos anticuerpos no se demostró un efecto importante sobre la función del FI, por lo que su papel patogénico en esta enfermedad aún no ha sido establecido, más aún cuando 2 de ellos además presentaban mutaciones en el gen de FH.
Otros anticuerposDebido al papel cada vez más reconocido, que tiene la vía alternativa en estas enfermedades, actualmente se están buscando autoanticuerpos frente a otras proteínas que puedan causar un mal funcionamiento de ella en algunos pacientes.
Siguiendo esta línea, nosotros hemos analizado la presencia de autoanticuerpos frente a FI, FB, C3 y properdina en muestras de alrededor de 85 pacientes con SHUa y 90 con glomerulonefritis (incluyendo C3G probadas), en los que no se han encontrado mutaciones ni otros anticuerpos asociados a estas enfermedades. También han sido estudiados aproximadamente 50 pacientes con una clara activación de la vía alternativa del complemento, pero sin ninguno de estos diagnósticos. Además, se ha analizado una cohorte de 100 pacientes con LES, como modelo de enfermedad autoinmune en la que el sistema del complemento está implicado y con una prevalencia mucho mayor que las C3G y el SHUa, para intentar buscar mecanismos patogénicos comunes.
En los grupos de pacientes con SHUa y glomerulonefritis estudiados, hemos encontrado un número variable de pacientes con anticuerpos frente a una o varias de estas proteínas, que son más frecuentes en las glomerulonefritis, aunque no en todos es evidente una activación de la vía alternativa.
De alguno de ellos se han realizado estudios funcionales en los que se ha comprobado que tienen un efecto discreto sobre la activación del complemento. En uno de estos casos, que además presentaba una mutación en heterocigosis en C3, se ha demostrado que la fracción IgG que contenía anticuerpos anti-FI, anti-FB, anti-C3 y antiproperdina activaba de forma especifica la vía alternativa en fase fluida, aunque no se pudo separar las distintas especificidades para comprobar si alguna en concreto era la responsable de esta activación36.
ConclusionesDesde hace tiempo se conoce la existencia de ciertos autoanticuerpos frente a componentes del sistema del complemento asociados a determinadas enfermedades, como es el caso del C3NeF y los anticuerpos anti-FH en las C3G y el SHUa, respectivamente. Estos anticuerpos alteran la regulación de la vía alternativa, al igual que ocurre con las mutaciones en las proteínas que componen esta vía y que han sido descritas asociadas a estas enfermedades.
Es en los últimos años cuando han aparecido descripciones de casos aislados de anticuerpos frente a otras proteínas o asociados a otras enfermedades. Este es el caso de los anti-FH y las glomerulopatías mediadas por complemento, aunque es necesario ampliar los estudios para comprobar su implicación en la enfermedad, sobre todo en los casos en los que aparecen junto al C3NeF.
En cuanto a los anticuerpos anti-FI, anti-C3, anti-FB y antiproperdina, es necesario realizar screenings sistemáticos en los pacientes para establecer su frecuencia real, aunque parecen ser más escasos que los autoanticuerpos anti-FH. Además, es necesario caracterizar cada anticuerpo detalladamente para poder determinar sus repercusiones en la regulación de la vía alternativa y, de esta manera, su papel en estas enfermedades.
La identificación de alguno de estos autoanticuerpos y de sus efectos sobre el complemento abre las posibilidades en cuanto a la elección del tratamiento en estos pacientes. Estos tratamientos pueden ir dirigidos a la inhibición del complemento o a la eliminación de estos autoanticuerpos mediante inmunosupresión o plasmaféresis.
Conceptos clave- •
La desregulación de la vía alternativa del complemento está implicada en la patogénesis de las C3G y del SHUa.
- •
Existen autoanticuerpos frente a diversos componentes del sistema del complemento que pueden alterar su funcionamiento.
- •
El C3NeF y los anticuerpos anti-FH tienen un claro papel en el desarrollo de C3G y SHUa, respectivamente.
- •
Existen autoanticuerpos frente a otras proteínas (FB, FI, C3, properdina), menos frecuentes, que pueden tener relevancia en estas enfermedades.
Este trabajo ha sido financiado por el Ministerio de Economía y Competitividad (SAF2012-38636), la Sociedad Española de Nefrología (Ayuda para la Investigación de la Fundación SENEFRO, 2013) y CIBERER (ACCI-2014).
Conflicto de interesesLos autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.



