





Complement system activation plays an important role in several renal pathologies, including antibody-mediated glomerulonephritis, ischaemia–reperfusion injury of transplanted kidneys or renal allograft rejection. Besides these conditions, alternative pathway abnormalities are directly involved in the pathogenesis of C3 glomerulopathies and atypical haemolytic uraemic syndrome. These abnormalities may be inherited or acquired, the latter as autoantibodies directed against the various components and regulators of the alternative complement pathway. The functional consequences of some of these antibodies and their association with these conditions are well known, whereas for other antibodies only isolated cases have been reported. This article describes the autoantibodies that target the alternative complement pathway proteins, their characteristics and their clinical relevance in renal pathologies.
La activación del sistema del complemento interviene en el desarrollo de varias enfermedades renales, como las glomerulonefritis mediadas por anticuerpos, el daño por isquemia-reperfusión en los trasplantes renales o el rechazo de los injertos. Además, alteraciones en la vía alternativa están directamente implicadas en la patogénesis de las glomerulopatías C3 y del síndrome hemolítico urémico atípico. Estas alteraciones pueden ser congénitas o adquiridas; estas últimas en forma de autoanticuerpos dirigidos contra los diversos componentes y reguladores de la vía alternativa del complemento. Las consecuencias funcionales de algunos de estos anticuerpos y su asociación con estas enfermedades se conocen desde hace tiempo, pero de otros solo existen descripciones de casos aislados. En este artículo, se describen los autoanticuerpos frente a proteínas de la vía alternativa del complemento, sus características y su implicación en la enfermedad renal.
The complement system is made up of a set of more than 50 soluble and membrane proteins that become sequentially activated in an enzyme cascade that has to be tightly regulated. Among the best-known functions of the complement system are the opsonisation and elimination of pathogens, as well as the removal of apoptotic debris and immune complexes from the circulation. Now, it is also known that the complement system plays an important role in the modulation of adaptive immune response and in many homeostatic processes unrelated to fighting against infections. Complement is also involved in a wide range of diseases, either as a result of excess, lack of activation or inadequate regulation.1
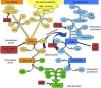
The complement system can be activated by three pathways: the classic, the lectin, and the alternative pathway (Fig. 1).
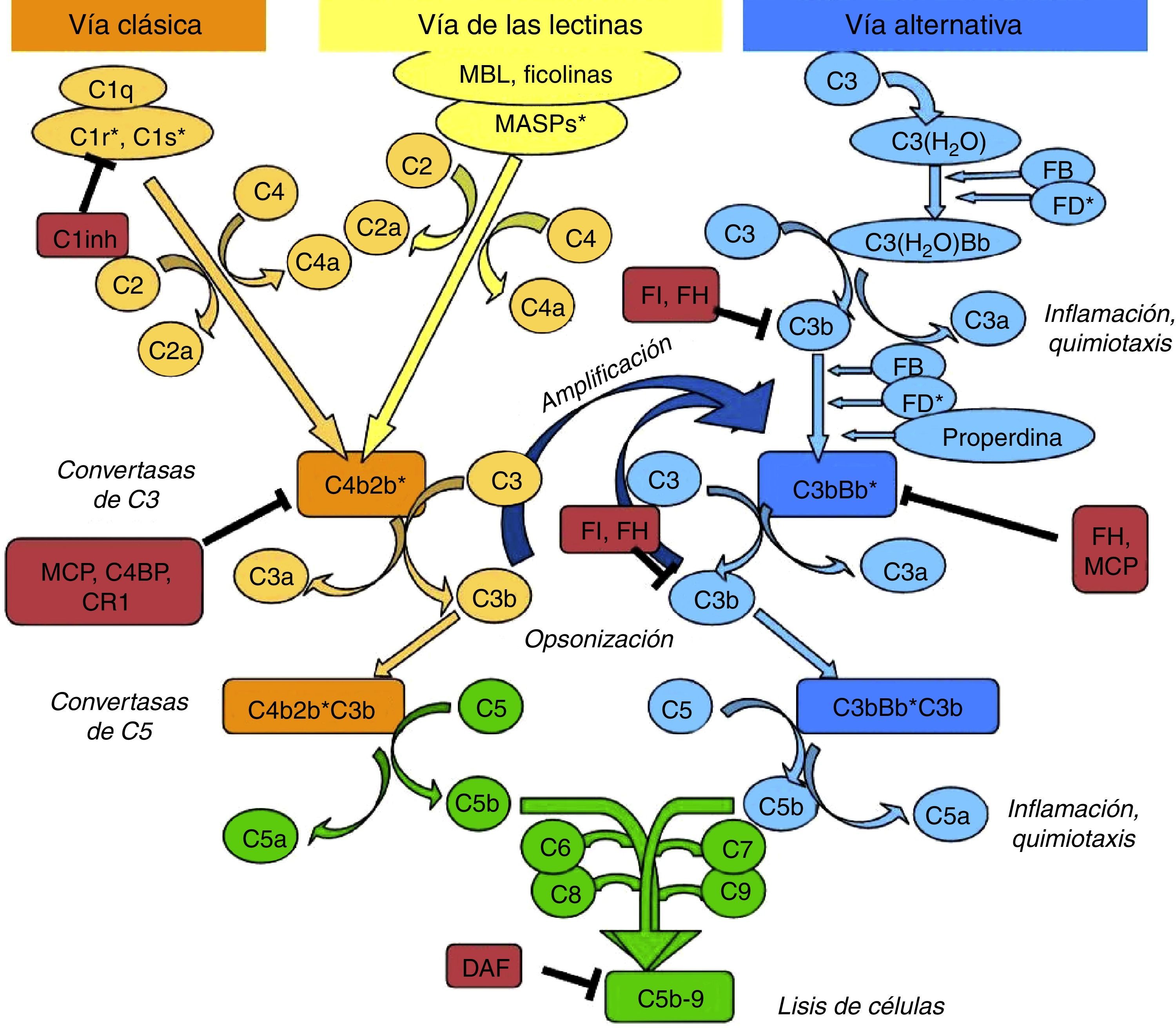
Diagram of activation of the complement system. The complement system can be activated by three pathways. Activation by any of them leads to the generation of C3 convertase (C4b2b* or C3bBb*) cleaving C3 into C3b and C3a. The C3b generated by any convertase can, in turn, form more alternative pathway convertase, through which amplification of complement activation occurs. The binding of a new molecule of C3b to the C3 convertases confers it the ability to cleave C5 into C5a and C5b. C5b initiates the terminal complement pathway which eventually leads to the formation of membrane attack complex (C5b-9) and lysis of the target cells. Complement activation is controlled at various levels by different regulatory proteins, both soluble and membrane-bound.
The classic pathway is activated essentially by the binding of C1q to antigen–antibody complexes, and the lectin pathway by the binding of MBL or ficolins to certain sugar residues on the bacterial cell surface. Activation of these two pathways results in the generation of a protein complex with enzymatic activity: the C3 convertase of the classic/lectin pathways (C4b2b) capable of cleaving C3 into C3a and C3b.
The alternative pathway is continuously and spontaneously activated by the hydrolysis of the thioester bond present in the C3 molecule. This C3(H2O) is able to bind to factor B (FB) which, once activated, forms the initial convertase of the alternative pathway which cleaves C3 resulting in the generation of C3a and C3b. C3b is deposited on cell surfaces and it is capable of forming new convertases by binding to FB (C3bBb). This favours the processing of more C3 molecules and the subsequent generation of more C3b, amplifying the complement cascade, regardless of the initial activation pathway. Properdin can either bind to these complexes and stabilise them, or bind directly to the surface of certain pathogens and thus serve as a platform for the formation of new convertases. When new C3b molecules bind to the C3 convertases, they form C5 convertases, which cleave C5, generating C5a, a potent anaphylatoxin, and C5b, which serves as an initiator for the formation of membrane attack complex when components of C6 to C9 are added.
To limit the activity of the convertases on the cell surfaces, there are soluble regulatory proteins (factor H [FH], factor I [FI]) and membrane proteins (MCP, DAF, CR1) which differentiate the body's own cells from pathogens and protect them from complement-mediated damage. The renal glomerulus is particularly vulnerable to inflammatory damage mediated by the complement system, which may be due to the presence of circulating activation fragments (systemic activation) or fragments produced in situ at the glomerular level (local activation).2 Although many diseases can be associated to the activation by different pathways, a common pathogenic mechanism is the damage caused by activation of C5.2
Complement activation is particularly important in the pathogenesis of atypical haemolytic uraemic syndrome (aHUS) and C3 glomerulopathy (C3G). In both cases, mutations are found in genes of the alternative pathway which are responsible for inadequate regulation of activation of that pathway. In addition to mutations, in some cases, autoantibodies are directed against the alternative pathway components,3 which can alter the function of its target proteins and interfere with complement regulation.
The autoantibodies to alternative pathway proteins described to date, their main characteristics and the clinical implications are described in detail (Table 1).
Antibodies against alternative pathway proteins in renal disease.
| Antibody | Epitopes | Effect | Disease (frequency) | Reference |
|---|---|---|---|---|
| C3NeF | C3 convertase (C3bBb). C3b and Bb | Stabilisation of convertase Massive consumption of C3 | DDD (80%), C3G (50%), acquired lipodystrophy, SLE. Healthy individuals | 4–6,10–13 |
| Anti-FH | SCRs 18–20 | Prevent regulation on cell surfaces | aHUS (10%) associated with FHR-1 deficiency | 16,19,21–23,25,26 |
| SCRs 1–5 | Interfere with FI cofactor activity | C3G | 28–33 | |
| Anti-FB/(anti-C3) | FB and Bb | Stabilisation of C3 convertase but inhibits activation of the terminal pathway. C3 consumption | DDD (1 case) | 14 |
| C3b and FB | Activation of convertase | DDD (2 cases) | 15 | |
| Anti-FI | FI | No effect | aHUS (<2%) | 34 |
| Anti-properdin/anti-C3/anti-FB/anti-FI | Properdin, C3, FB, FI | Activation of the alternative pathway in fluid phase | Lupus nephritis (one case) | 35 |
C3G: C3 glomerulopathy; C3NeF: nephritic factor; DDD: dense deposit disease; FB: factor B; FH: factor H; FI: factor I; SLE: systemic lupus erythematosus; aHUS: atypical haemolytic uraemic syndrome.
Nephritic factor (C3NeF) is an antibody that binds to the C3 convertase of the alternative pathway4 preventing its dissociation, increasing its half-life, and even preventing the action of regulatory proteins in some cases5 (Fig. 2). This stabilisation generally results in a massive systemic consumption of C3 and activation of the terminal pathway.6 Initially, it was considered that these autoantibodies recognised a neoepitope that was generated when the convertase enzymatic complex was assembled; later it was found that some antibodies are also able to recognise some of the convertase components alone (C3b, Bb).7
C3NeF. (A) Under normal conditions, C3 convertase is capable of cleaving C3 into C3b and C3a, but there are regulatory proteins (FH, MCP) that accelerate its dissociation and regulate spontaneous activation. (B) The existence of C3NeF stabilises the convertase, prevents the action of these regulators and allows it to remain active for a longer period of time being able to cleave more C3.
The C3NeF are a heterogeneous group of IgG or IgM antibodies that recognise different epitopes and cause different effects on complement activation.5,6,8 Because of this heterogeneity, detection and determination of their effects on complement regulation may sometimes be problematic.
C3NeF is strongly associated with dense deposit disease (DDD) and is found in approximately 80% of patients with this disease, but it is also present in 50% of patients with C3 glomerulonephritis and MPGN I and III8,9 and in acquired partial lipodystrophy,10 some cases of systemic lupus erythematosus11 and post-streptococcal glomerulonephritis.12 Despite the common association with DDD, it is not clear if the antibody is the origin of the disease or it is generated secondarily to the persistence of these neoepitopes since it is also found in healthy individuals13 and no correlation has yet been found with the patient's clinical expression.9 Whichever the case, continuous activation of C3 convertase, and in some cases C5 convertase, due to the presence of this antibody, contributes to DDD progression.
Anti-C3b and anti-FB antibodiesThe presence of antibodies that recognise individual convertase components has also been reported. First of all, an anti-FB antibody was found in a DDD patient with negative C3NeF by the traditional haemolytic assay. This antibody recognised the FB from serum and the Bb fragment when it was part of the C3 convertase, preventing both spontaneous and mediated by FH dissociation. It induced C3 consumption, but, unlike most C3NeFs, it inhibited the formation of C5 convertase.14
Later, in 2011, the simultaneous presence of anti-FB and anti-C3b antibodies was reported in two patients with DDD.15 These antibodies produced an increase in convertase activity and, as a result, increased generation of complement activation fragments. As in the case of C3NeF, it is unclear whether these autoantibodies are the cause of the disease or if they appear secondarily due to the increase in activation products circulating in plasma.
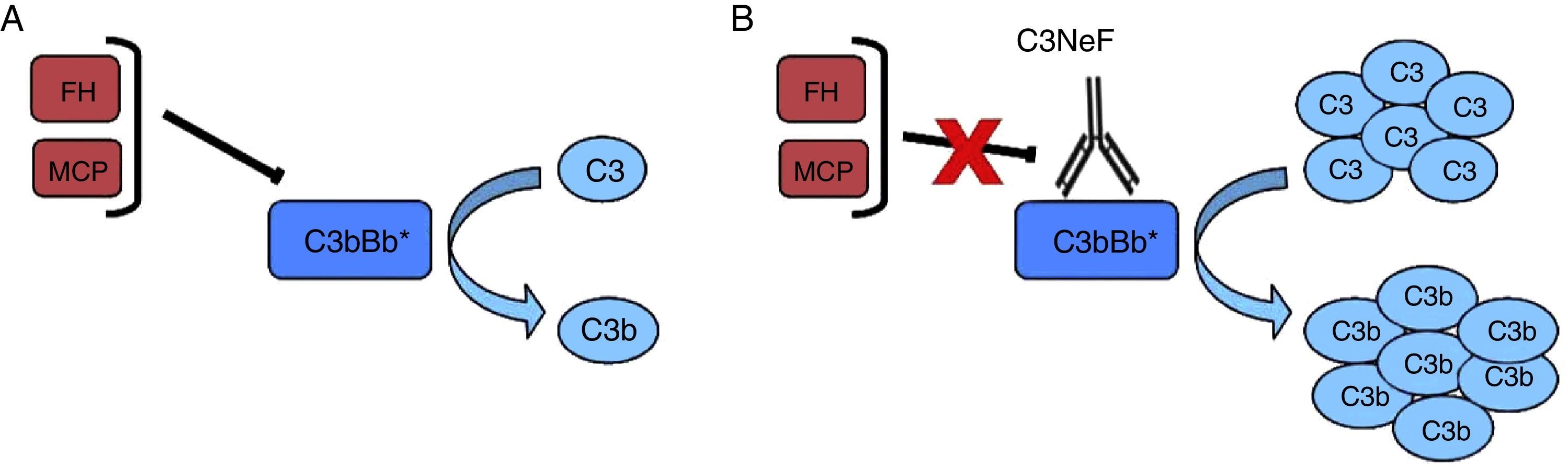
Anti-FH antibodiesThe anti-FH antibodies associated with aHUS have been known since 2005,16 and they are thought to be present in approximately 10% of cases in European population series17 and up to 56% in a large series of paediatric patients in India.18 The presence of anti-FH autoantibodies in aHUS is frequently associated with CFHR1 gene deletion.19–21 Initially, it was identified the epitope that these antibodies recognise in the C-terminal region of the FH molecule, the site of recognition and surface-binding domains 22,23 (Fig. 3). The SCRs 19–20 domains accumulate the majority of mutations of the FH gene associated to aHUS.24 More recently it has been found that, especially in the acute phase, these antibodies recognise other regions along the entire molecule, forming antigen–antibody complexes and blocking not only FH binding to cell surfaces, but also its activity as FI cofactor.25
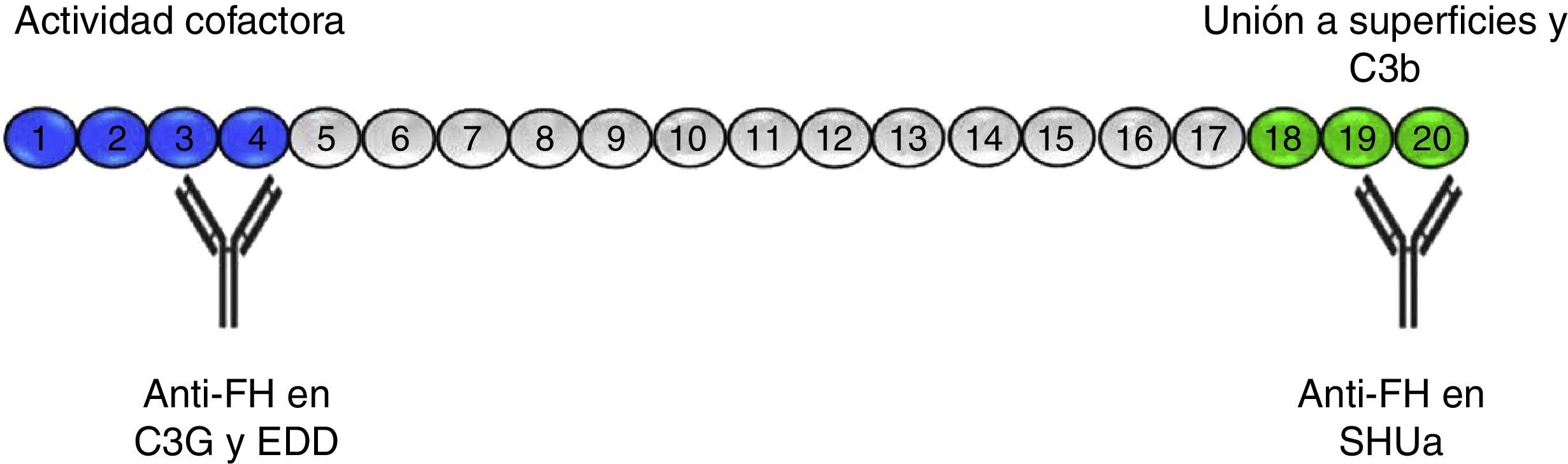
Anti-FH autoantibodies. FH is formed by 20 SCR domains. The N-terminus end of the protein (SCRs 1–4) is where the FI cofactor activity takes place and where the autoantibodies described in patients with C3G and DDD are directed. Through SCRs, 18–20 FH binds to cell surfaces and to the C3b that has been deposited on them. The anti-FH autoantibodies in aHUS recognise this region and prevent FH from binding to endothelial cells and the regulation of the alternative pathway at that level.
A recent study in 201526 using recombinant fragments with point mutations in FH SCRs 19–20 has more accurately defined the epitope that these antibodies recognise. In patients with FHR-1 deficiency, it has been found that anti-FH antibodies bind to a region in FH that acquires a similar configuration to FHR-1 after binding to certain ligands, including various bacterial proteins. Through this discovery, a model has been proposed to explain the role of the absence of FHR-1 in maintaining tolerance to FH.
From a cohort of about 400 patients diagnosed with aHUS, collected since 1999, 14 patients with anti-FH antibodies have been selected to study the characteristics of these autoantibodies. Except for one, all the patients had a childhood onset, which, in accordance with other series, corresponds to approximately 10% of the paediatric cases in this cohort.
The IgG subclass and light chain responsible for this activity have been characterised in this group of patients and the FH region recognised by the antibodies has been mapped using fragments of recombinant FH, including the functionally relevant regions of the protein. The anti-FH antibodies of the aHUS patients primarily recognise the C-terminal region, especially in patients with FHR-1 deficiency. The autoantibodies of patients without the FHR-1 deficiency, besides recognising the C-terminal region, also bind to other areas along the protein. In experiments with recombinant fragments containing the mutations, we obtained results consistent with Bhattacharjee et al., 26 which supports the proposed model on the generation of these autoantibodies, at least in patients with FHR-1 deficiency. We also analysed serial samples obtained during follow-up of patients to see if any changes occurred in the epitopes or affinity of these antibodies. We found that while there is heterogeneity among patients, the characteristics of their autoantibodies remain constant over time, although the total titre tends to decrease over time. All these results show a restricted oligoclonal response in the generation of these autoantibodies.27
The presence of anti-FH antibodies in patients with C3G is much less common than in aHUS, despite being first described in 1992.28 That first case was a “mini-antibody” formed by monoclonal lambda chain dimers which produced C3 consumption by inhibiting the regulatory function of FH. It was subsequently found that this antibody recognised SCR3, inhibiting FH ability to bind to C3b29 (Fig. 3). No more patients were reported until four years ago and cases are still very rare.30–33 In three of these cases, the FH region that the antibodies recognise has been identified, locating it at the N-terminal domain, responsible for regulatory activity. It was also found that the IgG fraction caused dysregulation of the alternative pathway31,33 and that, in one of them, the dysregulation was caused by the fact that they specifically blocked the cofactor activity of FH over FI.31
In the same year, it was published a series of 17 patients with C3G and anti-FH autoantibodies.34 These antibodies were directed against the N-terminal region of FH, and about one third of them altered the regulation in fluid phase, decreasing cofactor activity, but not the regulation on surfaces, as occurs in aHUS.
In these diseases, no association has been found with autoantibodies with deletion of CFHR 133 although it is common to detect anti-FH autoantibodies simultaneously with C3NeF and, to a certain extent, this makes it difficult to ascribe them a clear pathogenic role.
Although the presence of these antibodies in patients with glomerulonephritis is much less common than in aHUS, these antibodies should not be ruled out as the cause of complement deregulation since, in all cases where their functional implications have been studied, they have been found to be responsible for inhibiting the regulatory function of FH.
Anti-FI antibodiesIn a study of a cohort of 175 patients with aHUS, anti-FI antibodies were found in three of them.35 Binding to FI turned out to be specific in all three and circulating complexes were also found in the serum of these patients. However, functional tests with these antibodies showed no significant effect on FI function and their pathogenic role in this disease is therefore yet to be established, particularly when two of them also had mutations in the FH gene.
Other antibodiesDue to the increasingly widely accepted role the alternative pathway has in these diseases, there is now a focus on searching for autoantibodies to other proteins that might cause dysfunction of the alternative pathway in some patients.
In this light, we analysed the presence of autoantibodies to FI, FB, C3 and properdin in samples of around 85 patients with aHUS and 90 with glomerulonephritis (including confirmed C3G), in whom no mutations or other antibodies associated with these diseases had been found. We have also studied around 50 patients with clear activation of the alternative complement pathway, but with none of these diagnoses. In addition, a cohort of 100 patients with SLE were analysed as a model of autoimmune disease in which the complement system is involved and with a much higher prevalence than C3G and aHUS.
In the groups of patients with aHUS and glomerulonephritis, we found a variable number of patients with antibodies to one or more of these proteins, being more common in glomerulonephritis, although activation of the alternative pathway was not evident in every case.
Functional studies were carried out on some of them and the results were that they have a modest effect on complement activation. In one case, which also showed a heterozygous mutation in C3, it was demonstrated that the IgG fraction that contained anti-FI, anti-FB, anti-C3 and anti-properdin antibodies specifically activated the alternative pathway in fluid phase, although it was not possible to separate the different specificities to demonstrate whether any of them was responsible for this activation.36
ConclusionsThe existence of autoantibodies against components of the complement system associated with certain diseases, such as the C3NeF in C3G and anti-FH in aHUS, has been known for some time. These antibodies alter the regulation of the alternative pathway, as happens with mutations in the proteins integrating this pathway and that have been described in association with these diseases.
It is in recent years that reports have emerged describing isolated cases of antibodies against other proteins or associated with other diseases. This is the case of anti-FH and complement-mediated glomerulonephritis, although further studies are necessary to confirm their involvement in the disease, especially in cases that appear alongside C3NeF.
In the case of the anti-FI, anti-C3, anti-FB and anti-properdin antibodies, systematic screening should be performed in patients to establish their actual frequency, although they seem to be more rare than anti-FH autoantibodies. Additionally, each antibody needs to be characterised in detail to determine its impact on the regulation of the alternative pathway and, thus, its role in these diseases.
The identification of any of these autoantibodies and their effects on complement opens up possibilities in terms of the choice of treatment in these patients. These treatments could be targeted at complement inhibition or removal of these autoantibodies by immunosuppression or plasmapheresis.
Key concepts- •
Dysregulation of the alternative complement pathway is involved in the pathogenesis of C3G and aHUS.
- •
There are autoantibodies to various components of the complement system that can alter its function.
- •
C3NeF and anti-FH antibodies have a clear role in the development of C3G and aHUS, respectively.
- •
There are autoantibodies to other proteins (FB, FI, C3, properdin) which, although less common, may also be relevant in these diseases.
This work was funded by the Spanish Ministry of Economy and Competitiveness (SAF2012-38636), the Spanish Society of Nephrology (Fundación SENEFRO Aid for Research, 2013) and CIBERER (ACCI-2014).
Conflicts of interestThe authors declare that they have no potential conflicts of interests in relation to the contents of this article.
Please cite this article as: Nozal P, López-Trascasa M. Autoanticuerpos frente a proteínas de la vía alternativa del complemento en enfermedad renal. Nefrologia. 2016;36:489–495.



