


Pheochromocytomas and paragangliomas are tumours derived from neural crest cells, which can be diagnosed by biochemical measurement of metanephrine and methoxytyramine. Advances in genetic research have identified many genes involved in the pathogenesis of these tumours, suggesting that up to 35–45% may have an underlying germline mutation. These genes have a singular transcriptional signature and can be grouped into 2 clusters (or groups): cluster 1 (VHL and SHDx), involved in angiogenesis and hypoxia pathways; and cluster 2 (MEN2 and NF1), linked to the kinase signalling pathway. In turn, these genes are associated with a characteristic biochemical phenotype (noradrenergic and adrenergic), and clinical features (location, biological behaviour, age of presentation, etc.) in a large number of cases. Early diagnosis of these tumours, accompanied by a correct genetic diagnosis, should eventually become a priority to enable better treatment, early detection of complications, proper screening of family members and related tumours, as well as an improvement in the overall prognosis of these patients.
Los feocromocitomas y paragangliomas son tumores derivados de células de la cresta neural, que pueden ser diagnosticados mediante la determinación bioquímica de metanefrinas y metoxitiramina. Los avances en la investigación genética han permitido identificar múltiples genes implicados en la fisiopatogenia de estos tumores, de forma que hasta el 35-45% podrían tener una mutación germinal subyacente. Estos genes tienen una firma biológica de transcripción característica y se pueden agrupar en 2 grandes grupos (o clusters), el grupo 1 (VHL y SHDx), con implicación de la vía de la angiogénesis e hipoxia; y el grupo 2 (MEN2 y NF1), implicados en la vía de señalización de la cinasa. A su vez estos genes se asocian a un fenotipo bioquímico (adrenérgicos y noradrenérgicos), y presentación clínica (localización, comportamiento biológico, edad de presentación…) característicos en un número elevado de casos. Un diagnóstico precoz de estos tumores, acompañado de un correcto diagnóstico genético, debe ser una prioridad que permita un mejor tratamiento, la detección precoz de complicaciones, un correcto screening de familiares y de otros tumores relacionados, así como una mejoría en el pronóstico global de estos pacientes.
Pheochromocytomas (PH) and paragangliomas (PGL) are catecholamine-producing neuroendocrine tumours derived from neural crest chromaffin cells. The majority occur in the adrenal medulla (PH), but 20% are extra-adrenal.1
PH/PGL are a rare cause of secondary hypertension, with an incidence, in hypertensive patients, of 0.3–0.5%. The most common presentation is the combination of a varying degree of hypertension accompanied by paroxymal symptoms, either spontaneous or triggered by increased intraabdominal pressure or other types of stimuli. Paradoxically, some patients with PH/PGL may have episodes of orthostatic hypotension and even syncope, which are due to vasomotor receptor desensitisation or to a decrease in intravascular volume.2 At times, the clinical manifestations of these tumours are unrelated to changes in the blood pressure; so, in some cases, symptoms of other tumours associated with PH/PGL in the context of specific genetic mutations predominate (medullary thyroid cancer, renal cell cancer, haemangioblastomas, neurofibromas, neuroendocrine pancreatic tumours, gastrointestinal stromal tumour (GIST), pituitary tumours, etc.).3
The above symptoms do not occur in head and neck tumours; they tend to present as a non-catecholamine-producing growing mass.1,2
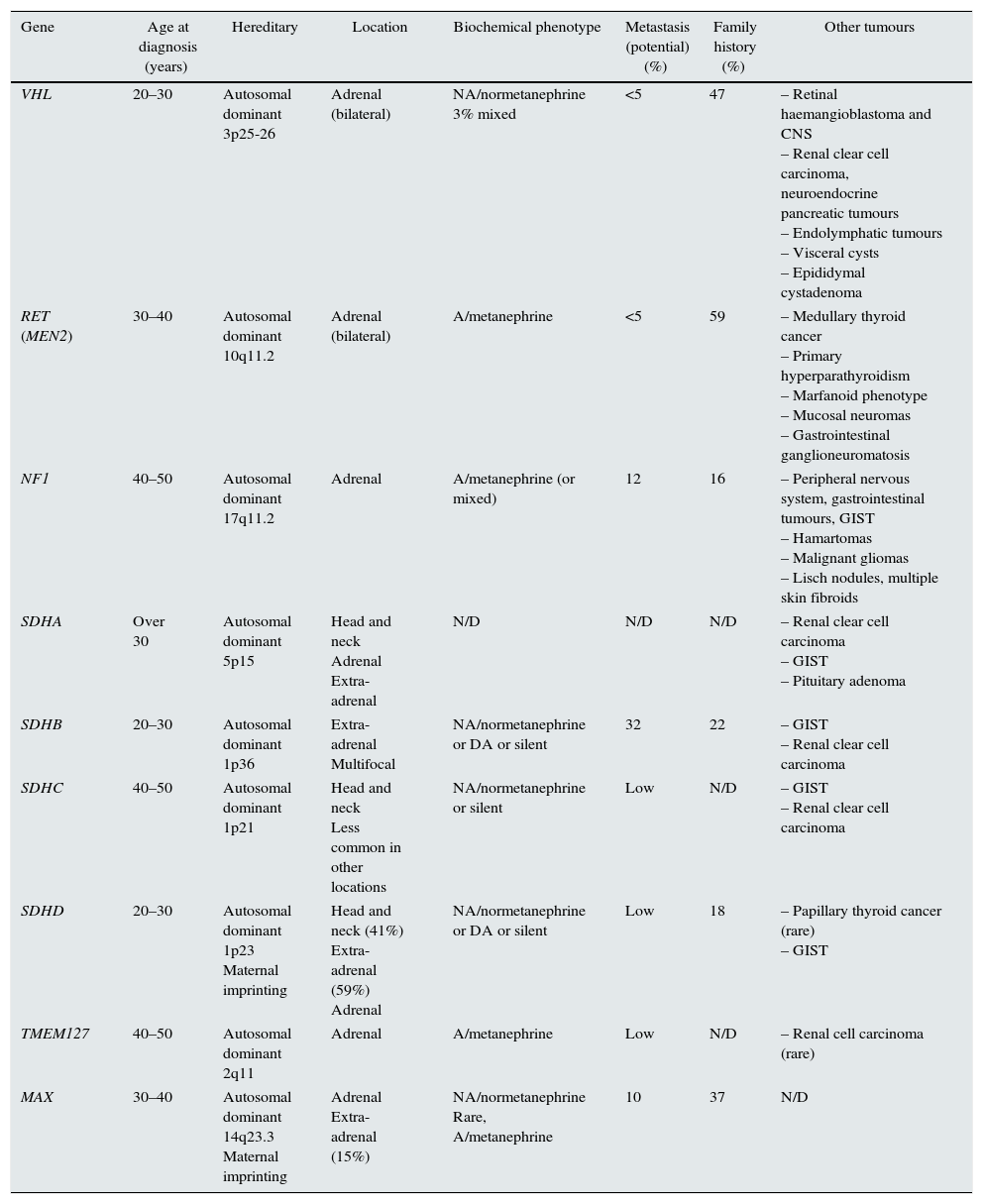
Depending on the series analysed, up to 35% of cases of PH and 15% of PGL are associated with a germline mutation; therefore, they can be considered genetically determined neuroendocrine tumours.4–6 Presently, there are at least 19 genes that are known to be related to the pathogenesis of these tumours (Table 1).2,3,5,7
Main features of clinical presentation, biochemical phenotype of the pheochromocytomas/paragangliomas and their genetic mutations.
| Gene | Age at diagnosis (years) | Hereditary | Location | Biochemical phenotype | Metastasis (potential) (%) | Family history (%) | Other tumours |
|---|---|---|---|---|---|---|---|
| VHL | 20–30 | Autosomal dominant 3p25-26 | Adrenal (bilateral) | NA/normetanephrine 3% mixed | <5 | 47 | – Retinal haemangioblastoma and CNS – Renal clear cell carcinoma, neuroendocrine pancreatic tumours – Endolymphatic tumours – Visceral cysts – Epididymal cystadenoma |
| RET (MEN2) | 30–40 | Autosomal dominant 10q11.2 | Adrenal (bilateral) | A/metanephrine | <5 | 59 | – Medullary thyroid cancer – Primary hyperparathyroidism – Marfanoid phenotype – Mucosal neuromas – Gastrointestinal ganglioneuromatosis |
| NF1 | 40–50 | Autosomal dominant 17q11.2 | Adrenal | A/metanephrine (or mixed) | 12 | 16 | – Peripheral nervous system, gastrointestinal tumours, GIST – Hamartomas – Malignant gliomas – Lisch nodules, multiple skin fibroids |
| SDHA | Over 30 | Autosomal dominant 5p15 | Head and neck Adrenal Extra-adrenal | N/D | N/D | N/D | – Renal clear cell carcinoma – GIST – Pituitary adenoma |
| SDHB | 20–30 | Autosomal dominant 1p36 | Extra-adrenal Multifocal | NA/normetanephrine or DA or silent | 32 | 22 | – GIST – Renal clear cell carcinoma |
| SDHC | 40–50 | Autosomal dominant 1p21 | Head and neck Less common in other locations | NA/normetanephrine or silent | Low | N/D | – GIST – Renal clear cell carcinoma |
| SDHD | 20–30 | Autosomal dominant 1p23 Maternal imprinting | Head and neck (41%) Extra-adrenal (59%) Adrenal | NA/normetanephrine or DA or silent | Low | 18 | – Papillary thyroid cancer (rare) – GIST |
| TMEM127 | 40–50 | Autosomal dominant 2q11 | Adrenal | A/metanephrine | Low | N/D | – Renal cell carcinoma (rare) |
| MAX | 30–40 | Autosomal dominant 14q23.3 Maternal imprinting | Adrenal Extra-adrenal (15%) | NA/normetanephrine Rare, A/metanephrine | 10 | 37 | N/D |
However, of all these genes, the most commonly involved in germline mutations in clinical practice are the Von Hippel–Lindau (VHL) genes and succinate dehydrogenase complex (SDHx) subunits D, B and C.4,5,8
Gene expression studies have evidenced possible common pathophysiological mechanisms for some of these genes, classifying them into two main groups or “clusters” according to the transcription pathways involved.
The first “cluster” is related to the activation of angiogenesis and pseudohypoxia pathways, and includes mutations in the VHL and SDHx genes. The second “cluster” consists of tumours associated with the neurofibromatosis type 1 (NF1) and RET proto-oncogene (multiple endocrine neoplasia type 2 or MEN2) gene mutation, which produce an alteration in the regulation of adrenergic metabolism, protein synthesis and the kinase-signalling pathway.6,7 These gene-expression profiles may be used to classify PH/PGL, assigning them to one of the two groups, each of which has specific therapeutic targets.4,8
In order to correctly identify a genetic mutation and therefore make an appropriate differential genetic diagnosis, the physician must obtain a precise personal and familial medical history, and take into account the following main aspects: age at presentation; tumour location; biochemical phenotype; and the presence of other associated tumours.5
Traditional approach to the diagnosis of PH/PGLBiochemical diagnosis: basic conceptsThe main indications for initiating a biochemical study to test for a possible PH/PGL are: clinical suspicion (paroxysmal hypertension, headache, diaphoresis, palpitations); unexplained variability in blood pressure; hypertensive crises to certain stimuli (anaesthetic induction, drugs, radiological contrast, surgery, exercise, childbirth); adrenal incidentalomas; and family history of PH/PGL. In young patients with hypertension and normal body weight, presence of diabetes is also a clinical indication for suspecting PH.9,10
PH and PGL synthesise, store and secrete the catecholamines dopamine (DA), adrenaline (A) and noradrenaline (NA). The catecholamines are partially or completely converted into the inactive metabolites metanephrine and normetanephrine by the catechol-O-methyltransferase present in the tumour cells.11,12
The release of catecholamines into the plasma may be paroxysmal or minimal, while release of metanephrines is more sustained over time, as the intra-tumour metabolism of catecholamines takes place regardless of their release and their release into plasma is therefore almost always continuous.5 Testing for metanephrines in plasma and urine is therefore more sensitive than testing for catecholamines, with fewer false negatives. In other words, normal levels of metanephrines in plasma or urine fairly reliably exclude the presence of a catecholamine-producing tumour, except in small tumours (<1cm), which do not release catecholamines, and the exceptional cases of tumours which only produce DA.5,11
In patients with slightly raised catecholamine or metanephrine levels, it is important to rule out false positives, and keep in mind that there are drugs that increase the release of catecholamines. Situations such as acute cardiovascular events can also raise catecholamine levels transiently (Table 2).2,13 Patients with chronic kidney disease, especially those on dialysis, often have elevated levels of plasma metanephrines in the absence of PH/PGL, so special caution must be taken in interpreting biochemical results in this population.14
Factors that can increase concentrations of catecholamines (false positives).
| Drugs that interfere with biochemical analysis and produce false positive results (suspend for at least 2 weeks) | Acute cardiovascular events |
|---|---|
| • Caffeine • β-Blockers • Sympathomimetics • Tricyclic antidepressants • MAOIs • Alpha-methyldopa • Levodopa • Paracetamol | • AMI • Acute pulmonary oedema • ACVA |
ACVA: acute cerebrovascular accident; AMI: acute myocardial infarction; MAOI: monoamine oxidase inhibitor.
Source: Adapted from Kronenberg et al.39
False positive patients usually have higher increases in catecholamine levels as compared to metanephrine due to sympathoadrenal activation, so a normetanephrine/noradrenaline (NA) ratio<0.52 and metanephrine/adrenaline (A) ratio<4.2 points to sympathoadrenal activation.11 The clonidine suppression test can be a method to distinguish true positives from false positives with borderline NA levels. Clonidine is an α2-adrenergic receptor agonist that inhibits the release of neuronal NA in patients without PH/PGL, but not in the presence of autonomous tumour secretion of catecholamines. This test has a specificity of 100%, with a sensitivity of 97%, but so far has not been validated in any prospective study.2,5
To distinguish between the different inherited forms, it is important to note that NA is only converted to A by phenylethanolamine-N-methyltransferase (PNMT) in the chromaffin cells of the adrenal medulla. Thus, A is produced almost exclusively in adrenal tumours, whereas extra-adrenal tumours produce mainly NA. This is due to the fact that cortisol is able to induce the synthesis of PNMT in the adrenal glands, and because the accumulation of succinate in tumour tissue causes hypermethylation of the PNMT promoter gene and prevents its synthesis.4
Plasma and urine concentrations of metanephrines may be normal in some cases of large, extra-adrenal abdominal, metastatic PGL or PGL with the SDHB mutation (usually tumour cells that have lost their ability to synthesise by dedifferentiation).15 This may be due to a defect in the synthesis of catecholamines due to the absence of tyrosine hydroxylase (TH), a rate-limiting enzyme in catecholamine synthesis.16
Tumours producing only DA are rare, and are usually extra-adrenal PGL, so testing for DA or its metabolite (methoxytyramine) would be indicated in patients with an atypical presentation, in whom suspicion of PGL is high and metanephrine levels are normal.11,16
Head and neck PGL, which tend to be biochemically silent, may also be positive for TH, which converts tyrosine into dihydroxyphenylalanine (dopamine precursor), such that up to 23% of cases may have increased urinary excretion of 3-methoxytyramine; and in this type of tumour, it may be a better marker than detecting urine DA.17
Location: imaging techniquesOnce the biochemical diagnosis has been made, the tumour has to be located. Computed tomography or magnetic resonance imaging are the initial imaging tests of choice in the diagnostic process; these tumours have their own characteristics.18,19 It should be remembered that up to 25% of all PH are discovered incidentally and, depending on the series, up to 8% of adrenal incidentalomas are PH.7,20
However, if the tumour is small, in an unusual location or there are post-surgical changes, these tests may not be able to detect it.
Complementing the anatomical diagnosis with a functional imaging test is therefore recommended, especially in suspected extra-adrenal tumours, genetic syndrome (characteristic phenotype or young age), large tumours (>3cm) or bilateral involvement.21
The most commonly used functional imaging tests are scintigraphy with metaiodobenzylguanidine (MIBG) and PET with 18F-fluorodopamine (18F-FDA) or 18F-fluorodihydroxyphenylalanine (18F-FDOPA).
Functional imaging tests are based on transport of radiopharmaceuticals through the catecholamine transport systems located on the cell membrane; the most specific is the NA membrane transporter (NET), which is responsible for the reuptake of NA/DA.
MIBG is a guanidine analogue with structure similar to NA, which is captured through the NET transporter and concentrated by functioning chromaffin tissues. In addition, PH/PGL can capture and decarboxylate amino acids such as dihydroxyphenylalanine (DOPA). Thus, 18F-FDOPA, which is a catecholamine precursor, is captured by the amino acid transporter (LAT) of chromaffin cells and it is converted into 18F-FDA21 by the aromatic l-amino acid decarboxylase enzyme, which catalyses a rate-determining step in catecholamine synthesis. 18F-FDA, which is similar to DA, is captured by the chromaffin cells by the NET transporter, but with a greater affinity than MIBG, so PET 18F-FDA has the highest sensitivity and specificity in patients with biochemical diagnosis of PGL, independently of their genetic status.14 In general, 18F-FDA or 18F-FDOPA may be the best choice to locate the primary tumour or detect metastatic or multifocal disease.16 The main problem with these techniques is that they are only available in certain centres.16,22
In general, MIBG has a sensitivity of 80% and a specificity of 99%. However, 123I-MIBG sensitivity drops to 56–70% for PGL. It is not, therefore, the best choice for studying extra-adrenal lesions, metastasis, recurrence, or suspected hereditary syndromes (especially in the case of mutations in the SDHx – mainly mutations in SDHB – and VHL genes). It is also not the best test to study head and neck PGL.14 However, if a non-metastatic sporadic PH is suspected, 123I-MIBG can be as sensitive as PET and superior to 111In-DTPA-pentetreotide (Octreoscan) in locating the tumour.21
Other causes of false negatives with MIBG must also be taken into account, such as small lesions, necrosis, poorly differentiated tumours (loss of NET), drugs that act on the adrenergic system with intense adrenal uptake. Thus, if metanephrine levels remain elevated after a surgery of PH, a 123I-MIBG scan should be considered to rule out metastases, since the first scan may not have detected small metastases due to increased metabolic activity of the primary tumour.21–24
For cases in which malignancy, multifocal disease or the presence of SDHx-type germline mutations is suspected, performing the functional study with tracers other than 123I-MIBG is recommended. The sensitivity of these tracers (18F-FDA, 18F-FDOPA) ranges from 81% to 100%.14,21,22
If the PET study is also negative, we are probably dealing with an unusual type of PH – in which the tumour cells do not express the NET transporter system or may not have many catecholamine storage granules – or a malignant PH. In this case, a test with non-specific ligands, such as those related to the somatostatin receptor (SSTR), may be indicated. Scintigraphy with 111In-pentetreotide can detect lesions that do not take up 123I-MIBG or 18F-FDA.3,22 Indium-111 (111In)-labelled pentetreotide binds specifically to the SSTRs expressed in the cell membranes, especially subtypes 2 and 5. SSTRs are expressed in many cells of neuroendocrine origin and also in tumours deriving from these cells. However the SSTR expression may be variable which may affect the sensitivity of the technique.21
Approaching the diagnosis from a genetic point of viewFamily history: general inheritance patternsUp to 35–45% of cases may have an underlying germline mutation.3,6,7 Many of the genes involved in PH/PGL have incomplete penetrance and variable phenotypic expression, and therefore only about 10% of PH/PGL has a family history of PH.
Most hereditary PH/PGL transmission is autosomal dominant by way of a “second hit” mechanism (loss of wild-type allele in the tumour), but in the case of PH/PGL due to mutations of the proto-oncogene RET and hypoxia inducible factor 2A (HIF2A), they only need the mutation in one allele to be activated (no need for “second hit”).4,5,8 Patients with PH/PGL associated with mutations in the RET, VHL and NF1 genes usually have a typical family history. However, it should be noted that approximately 50% of cases with MEN2B syndrome or changes in the NF1 gene have a de novo mutation. Furthermore, patients with mutations in the SDHx present with family history in only 30% of cases, due to incomplete penetrance of the syndromes; therefore, a negative family history does not exclude the presence of a hereditary tumour. In that regard, we have to bear in mind that genes like SDHD, SDHAF2 and MYC-associated factor X (MAX) have maternal imprinting, i.e. only paternal transmission leads to the disease.4,5
In children, an underlying germline mutation is identified in up to 70% of sporadic cases.3 If there is no characteristic phenotype or family history of MEN2, the most likely diagnosis is a mutation in VHL or, an SDHx mutation.9,25 In the paediatric population, there is also a greater likelihood of larger, extra-adrenal tumours, the existence of synchronous tumours and malignancy.26
Genetic diagnosis and biochemical phenotypeThe genetic diagnosis (Table 1) can be oriented by the biochemical phenotype of PH/PHL, which would include the following categories: adrenergic tumours (A, metanephrine); noradrenergic tumours (NA, normetanephrine); mixed tumours; and dopaminergic tumours (DA, methoxytyramine). For mixed tumours, and to facilitate diagnosis, those producing NA and A are considered adrenergic, while tumours producing NA and DA would be dopaminergic.5
VHL tumours are almost always located in the adrenal gland (PH) they have an NA- and normetanephrine-producing phenotype, and only 3% have elevations of A or metanephrine.27
In cases of PH exclusively producing NA, presence of MEN2 syndrome is unlikely (since in that case they would be producers of A and metanephrine).11,28 This is because the PH in patients with MEN2 (unlike VHL) express PNMT and have greater TH activity.27
The chromaffin tumours due to mutations in the SDHx produce mostly NA/normetanephrine.28 Elevation of DA, and of its metabolite methoxytyramine, is also observed in some cases of SDHx.5,11–13,29 Methoxytyramine may be the only marker in cases of tumours exclusively producing DA, being a predictive marker for malignancy in these tumours that tend to be clinically silent.11–13 Metastatic tissue lacks enzymes for the synthesis of catecholamines, so high levels of DA, DOPA and methoxytyramine could be a marker for metastatic disease.11 Increased levels methoxytyramine in plasma may even be a more sensitive marker of tumour production of DA and of metastatic dissemination than the plasma or urinary DA levels.30
Mutations in RET, transmembrane protein 127 (TMEM127), and NF1 genes express an A/metanephrine secretory phenotype, whereas mutations in MAX and HIF2A express a noradrenergic phenotype.4
To improve the genetic registers and better define the incidence and prevalence of these syndromes, patients should be included in registries. In Spain, the reference centre for genetic study of these tumours is coordinated by Dr Mercedes Robledo (mrobledo@cnio.es) at the National Cancer Research Centre (CNIO) in Madrid.
Genetics and functional imagingFor VHL, NF1 or RET mutations, it is preferred the used of 18F-FDA or 18F-FDOPA. In the case of VHL, up to 50% of PH tends to be bilateral and 18F-FDA is superior to MIBG due to the low expression of NET in these cases.22,31,32
If SDHx mutations are suspected, use of 18F-FDA (18F-FDOPA in the case of head and neck PGL), 18F-FDG or 111In-DTPA-pentetreotide is recommended.
In general, tumours associated with certain SDHx mutations (SDHC, SDHD and SDHAF2) are located in the head and neck. If high levels of catecholamines are found, imaging studies should be performed to identify another tumour – usually located in the abdomen or pelvis.33SDHB mutations are associated with a higher rate of malignancy (70%) and extra-adrenal tumours. PH may occur in 25% of the cases and the risk of bilateral involvement is low.3,6,29,34SDHC mutations may be present in up to 4% of cases of head and neck PGL, but it is very rare for them to be associated with PH.5,8SDHD mutation is typically associated with parasympathetic head and neck PGL and multifocality; adrenal tumours with SDHD mutations are not usually bilateral.7,15,34
The greater utility of 18F-FDG for functional diagnostics in the case of SDHx and VHL mutations and the exact mechanism involved in 18F-FDG uptake are not fully understood. It is known that these tumours have increased expression of genes associated with angiogenesis and hypoxia, such that there is increased activation of aerobic glycolysis, with increased phosphorylation of glucose by hexokinases, leading to greater accumulation of 18F-FDG.35
PET with 18F-FDG is currently being used more often to detect the presence of metastatic disease, tumour recurrence and response to chemotherapy. However, it is not indicated for initial location because, depending on the location and the genetics, it has lower sensitivity and specificity than other tracers (sensitivity of 62% in SDHB-negative vs 83% in SDHB-positive).36 In cases of aggressive PH, which can be negative with the tracers mentioned above, 18F-FDG may be useful in diagnosing the location of metastases.4,36
ConclusionsPH/PGL are neuroendocrine tumours with a significant genetic basis. Advances in genome studies have led to better characterisation of these tumours and this knowledge needs to be applied in routine clinical practice. Therefore, priority must be given to genetic study according to clinical data, family history, biochemical phenotype and tumour location.
In general, we would recommend genetic study in the case of PGL, bilateral adrenal PH, unilateral adrenal PH and family history of PH/PGL, unilateral adrenal PH in people under the age of 30 and in all cases suggestive of a familial syndrome.9,21
- •
35% of PH and 15% of PGL are associated with a germline mutation.
- •
A negative family history is not uncommon, especially in cases of SDHX mutations, due to incomplete penetrance and variable phenotypic expression of these genes.
- •
Most hereditary PH/PGL have a specific biochemical phenotype. Methoxytyramine may be useful in the study of biochemically silent or DA-producing PGL.
- •
The algorithm shown in Fig. 1 is a suggested genetic diagnostic approach in these patients.

The authors declare that there are no conflicts of interest that might be perceived as detrimental to the impartiality of the published research.
Please cite this article as: Cano Megías M, Rodriguez Puyol D, Fernández Rodríguez L, Sención Martinez GL, Martínez Miguel P. Feocromocitoma-paraganglioma: del diagnóstico bioquímico al genético. Nefrología. 2016;36:481–488.



