
Two types of early childhood hyperkalemia had been recognized, according to the presence or absence of urinary salt wasting. This condition was attributed to a maturation disorder of aldosterone receptors and is characterized by sustained hyperkalemia, hyperchloremic metabolic acidosis (MA) due to reduced ammonium urinary excretion and bicarbonate loss, and normal creatinine with growth delay. We present 3 patients of the type without salt wasting, which we will call transient early-childhood hyperkalemia (TECHH) without salt wasting, and discuss its physiopathology according to new insights into sodium and potassium handling by the aldosterone in distal nephron. In 3 children from 30 to 120-day-old admitted with bronchiolitis and growth delay hyperkalemia was found in routine laboratory. Further studies revealed a normal creatinine with inappropriately normal or low fractional excretion (FE) of potassium, accompanied by inadequately normal serum aldosterone and plasma renin activity for their higher plasma potassium levels, but without urine salt wasting. They also presented hyperchloremic MA with FE of bicarbonate 0.58%–2.2%, positive urinary anion gap during MA and normal ability to acidify the urine. Based on these findings a diagnosis of TECHH without salt wasting was made and they were treated sodium bicarbonate and hydrochlorothiazide with favorable response. The condition was transient in all cases leading to treatment discontinuation. Given that TECCH without salt wasting is a tubular disorder of transient nature with mild symptoms; it must be keep in mind in the differential diagnosis of hyperkalemia in young children.
Se reconocen dos variedades de hiperpotasemia temprana de la infancia (del inglés Early childhood hyperkalemia) según la presencia o no de pérdida salina urinaria. Se trata de una entidad atribuida a un desorden madurativo en los receptores de aldosterona caracterizada por hiperpotasemia, acidosis metabólica (AM) hiperclorémica por diminución de la eliminación de amonio y bicarbonaturia, y creatinina normal con retraso de crecimiento. Presentamos 3 pacientes de la forma con ausencia de pérdida salina, a la que denominaremos hiperpotasemia transitoria del lactante (HTL) sin pérdida salina, y discutimos su fisiopatología en relación a los nuevos conocimientos en el manejo tubular del sodio y el potasio por la aldosterona. En 3 pacientes de entre 30 y 120 días de edad con bronquiolitis y retraso de crecimiento se encontró hiperpotasemia en laboratorio de rutina. Presentaban creatinina normal, excreción fraccionada (EF) de potasio disminuida o inapropiadamente normal junto a niveles de aldosterona y renina plasmática inadecuadamente normales para el estado de hiperpotasemia, pero sin pérdida salina. También cursaban con AM hiperclorémica con bicarbonaturia (EF bicarbonato 0,58%–2,2%), anión restante urinario positivo durante AM y capacidad normal para acidificar la orina. En base a estos hallazgos se diagnosticó HTL sin pérdida salina y se trataron con bicarbonato de sodio e hidroclorotiazida con buena respuesta. El cuadro fue transitorio permitiendo la suspensión del tratamiento. Dado que el HTL sin pérdida salina es un desorden tubular transitorio con síntomas leves debe tenerse presente en el diagnóstico diferencial de hiperpotasemia en niños pequeños.
In a Nephrology Forum on renal tubular acidosis in 1981, McSherry talked about a group of 13 infants with a condition he called early-childhood hyperkalaemia.1 It was characterised by hyperkalaemia and hyperchloraemic metabolic acidosis (MA) due to decreased ammonium excretion and bicarbonate in urine. Their urine also had decreased or inappropriately normal fractional excretion (FE) of potassium (K+) for the elevated serum K+ values and normal FE of sodium (Na+), with elevated or inappropriately normal plasma aldosterone and renin activity. This syndrome was attributed to a probable maturational disorder in the number or function of mineralocorticoid receptors in the distal tubule, leading to resistance to the action of aldosterone in the distal nephron.1–3 However, this theory did not explain the absence of salt wasting in these patients.4 In 1986, Appiani et al. published for the first time the cases of five patients with early-childhood hyperkalaemia, but with a different phenotype, as unlike the previous cases, they did have urinary salt wasting.2 To our knowledge, no new cases have been reported since then. We now present three additional patients with the syndrome described by McSherry,1 which we refer to throughout the manuscript as “transient early-childhood hyperkalaemia without salt wasting” (TECH without salt wasting) to differentiate it from the variant described by Appiani et al.2 We provide a new pathophysiological approach based on advances in our understanding of the channels and transporters that regulate aldosterone-mediated Na+ and K+ transport in the distal tubule.
MethodsBlood gases were determined in arterialised capillary samples. The pH and pCO2 in blood and urine were determined by an ABL 520® analyser, while Na+ and K+ were measured with the ion-selective Radiometer®. Urine samples were collected by catheter and kept anaerobically in a sealed syringe until measurement of urinary pH, pCO2 and bicarbonate (HCO3−) (pHu, pCO2u and HCO3−u, respectively). The HCO3−u was calculated with the Henderson-Hasselbalch equation, using the solubility constant of pCO2 in blood (α=0.00301) and a blood pK of 6.1.5 Urinary ammonia was inferred by the urine anion gap (AG), compared to spontaneous MA or during the furosemide test.6 The FE of Na+, K+ and HCO3− were calculated with the formula (U/P of the substance)/(U/P creatinine)×100; where U and P represent the concentrations in urine and plasma, respectively. Creatinine was determined with the Jaffé method, while plasma renin and aldosterone were measured by radioimmunoassay in samples collected at nine o'clock in the morning. Hyperchloraemia was defined as a chloride (Cl–) concentration >75% of the concentration of Na+.7 The plasma AG was calculated with the formula Na+ – (Cl– + HCO3−), considering a normal value 12±2, and the urinary AG with the formula (Na+ + K+) – Cl−, considering that any negative value suggests normal ammonium excretion, while any positive value reflects a decrease in excretion. The alkali loading test to determine the difference in blood and urine pCO2 (U-B pCO2) was adapted for use in children and a difference ≥20mmHg was considered normal.8 Calciuria was determined in isolated urine, averaging three determinations, dividing the calcium concentration with the creatinine concentration and considering a normal value to be <0.8.9 The transtubular potassium gradient was calculated with the formula (U/P of K+)/(U/P osmolality)×100; osmolality was measured with a vapour pressure osmometer (Wescor®), with the value in infants with hyperkalaemia expected to be >5.3 The normal electrolyte and creatinine values were the usual ones for the age groups studied.9–12 Blood pressure was recorded with an oscillometric device. Three consecutive readings were made, we considered the value resulting from the average of the three measurements.13
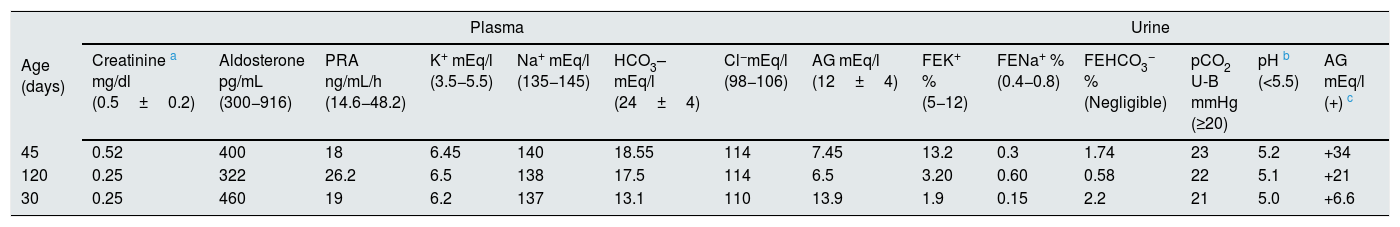
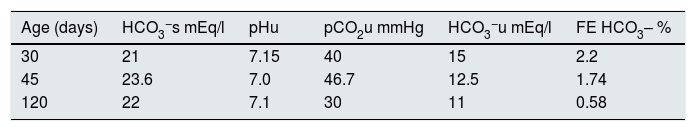
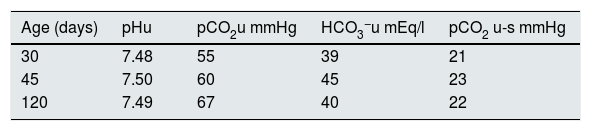
PatientsThree patients (two male) aged 30, 45 and 120 days, respectively, hospitalised for bronchiolitis syndrome, had laboratory parameters compatible with TECH without salt wasting. Two of the patients had no history of perinatal problems, while the 120-day-old boy was born at 30 weeks' gestation with a weight of 1.735kg (adequate weight for gestational age) and had had hyaline membrane disease requiring mechanical ventilation for seven days. During their hospital stay, sustained hyperkalaemia was found in all cases, accompanied by normal natraemia and hyperchloraemic MA. None of them was receiving medication or had any other external factors likely to cause elevation of serum K+. Furthermore, the disorder persisted despite treatment with β2 agonists, indicated by the respiratory symptoms. All three patients had normal blood pressure: the 30-day-old 70/50mmHg; the 45-day-old 75/55mmHg; and the 120-day-old 70/50mmHg. Blood AG, lactic acid, ammonium and creatinine values were normal in all patients. Plasma renin activity and serum aldosterone were inappropriately normal in all three cases. In addition, all three had low or inappropriately normal FEK+ for the degree of hyperkalaemia, with normal FENa+ (Table 1). The transtubular potassium gradient was only determined in the 120-day-old patient and was 2.66. Calcium excretion was normal, with values of 0.66, 0.5 and 0.72 in the 30-, 45- and 120-day-old patient, respectively, and urinary infection was ruled out by negative urine culture. Renal ultrasounds were normal, with no findings compatible with urological disorders such as dilated urinary tract and/or bladder disease. Congenital adrenal hyperplasia was ruled out based on the finding of normal external genitalia along with normal serum levels of 17-OH progesterone, 18-OH corticosterone, ACTH, cortisol and dehydroepiandrosterone sulfate. The urinary AG was positive, becoming negative after administration of furosemide; while the FE HCO3− was elevated after normalising blood values with alkalis (Table 2). In addition, compromise of hydrogen (H+) secretion through the distal tubule was ruled out as cause of the MA, as the U-B pCO2 was normal (>20mmHg) (Table 3).
Metabolic profile in three patients with transient early-childhood hyperkalaemia without salt wasting.
| Age (days) | Plasma | Urine | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Creatinine a mg/dl (0.5±0.2) | Aldosterone pg/mL (300−916) | PRA ng/mL/h (14.6−48.2) | K+ mEq/l (3.5−5.5) | Na+ mEq/l (135−145) | HCO3– mEq/l (24±4) | Cl−mEq/l (98−106) | AG mEq/l (12±4) | FEK+ % (5−12) | FENa+ % (0.4−0.8) | FEHCO3− % (Negligible) | pCO2 U-B mmHg (≥20) | pH b (<5.5) | AG mEq/l (+) c | |
| 45 | 0.52 | 400 | 18 | 6.45 | 140 | 18.55 | 114 | 7.45 | 13.2 | 0.3 | 1.74 | 23 | 5.2 | +34 |
| 120 | 0.25 | 322 | 26.2 | 6.5 | 138 | 17.5 | 114 | 6.5 | 3.20 | 0.60 | 0.58 | 22 | 5.1 | +21 |
| 30 | 0.25 | 460 | 19 | 6.2 | 137 | 13.1 | 110 | 13.9 | 1.9 | 0.15 | 2.2 | 21 | 5.0 | +6.6 |
AG: anion gap; FE: fractional excretion; PRA: plasma renin activity; U-B pCO2: difference in blood and urine pCO2.
The normal values for age are shown in brackets.
Determination of bicarbonate and fractional bicarbonate excretion in three patients with transient early-childhood hyperkalaemia without salt wasting.
| Age (days) | HCO3−s mEq/l | pHu | pCO2u mmHg | HCO3−u mEq/l | FE HCO3– % |
|---|---|---|---|---|---|
| 30 | 21 | 7.15 | 40 | 15 | 2.2 |
| 45 | 23.6 | 7.0 | 46.7 | 12.5 | 1.74 |
| 120 | 22 | 7.1 | 30 | 11 | 0.58 |
Values after administration of alkali loading.
s Determination in serum.
u Determination in urine.
Alkali loading test to determine the difference between urine and blood pCO2 (U-B pCO2) in three patients with transient early-childhood hyperkalaemia without salt wasting.
| Age (days) | pHu | pCO2u mmHg | HCO3−u mEq/l | pCO2 u-s mmHg |
|---|---|---|---|---|
| 30 | 7.48 | 55 | 39 | 21 |
| 45 | 7.50 | 60 | 45 | 23 |
| 120 | 7.49 | 67 | 40 | 22 |
Values after administration of alkali loading.
s Determination in serum.
u Determination in urine.
The hyperkalaemia was treated with cation exchange resins 1g/kg every 8−12h and furosemide 1mg/kg every 6h. The furosemide was later replaced by hydrochlorothiazide, to prevent loop diuretic-related hypercalciuria. However, just recently we were able to normalise the serum concentration of K+, and the MA, after the addition of sodium bicarbonate orally at 2mEq/kg/day. During follow-up, treatment was suspended at times, but with reappearance of the metabolic disorders, until definitive resolution was verified at 6 months in the 30-day-old patient, 7 months in the 120-day-old and 11 months in the 45-day-old. Over the course of their recuperation, weight and height recovery was observed in all three cases. The 30-day-old patient weighed 3.6kg (3rd–10th percentile) at diagnosis and measured 52cm (10th percentile), reaching a weight of 7.9kg and a height of 67cm at six months of age, both in the 25th–50th percentile. In the case of the 45-day-old, who at the beginning weighed 3.6kg and measured 50cm, both below the 3rd percentile, by the age of 11 months weighed 9.4kg (50th percentile) and measured 74cm (25th–50th percentile). Similarly, the 120-day-old weighed 3.25kg (3rd percentile) and measured 52cm (3rd–10th percentile) at diagnosis, but had reached 5.5kg and a height of 60cm, both in the 25th–50th percentile, at the age of seven months.
DiscussionIn this study, we describe in detail three patients with TECH without salt wasting with a metabolic pattern similar to those discussed by McSherry.1 Our patients had MA induced by the same hyperkalaemia, as it inhibits ammoniagenesis, leading to a decrease in the urinary excretion of ammonia.14 Moreover, the elevated K+ concentration was also responsible for the bicarbonate in urine which, although showing a lower level of excretion than that observed in proximal renal tubular acidosis (pRTA), was higher than that in healthy infants and children in whom HCO3− secretion is negligible.15 Therefore, persistent MA caused by the two mechanisms mentioned could have brought about the growth retardation observed in these children. H+ secretion by the cortical collecting duct (CCD) was adequate based on the fact that the U-B pCO2 was normal, this being the most sensitive marker in H+ secretion by the intercalated α cells, and so distal renal tubular acidosis (dRTA) could be ruled out. It is worth mentioning that while in cases caused by a gradient defect due to increased permeability of the luminal membrane of the CCD, as can occur in amphotericin toxicity, the U-B pCO2 is normal (≥20mmHg) but the urinary pH remains higher than 5.5 compared to MA,16 in our patients, the pH fell below 5.5, making it unlikely that this mechanism was responsible for their acidosis. We were able to differentiate our patients' conditions from pseudohypoaldosteronism type 13 or being secondary to urinary tract infection and/or malformations17 by the absence of salt wasting from the kidney and hyponatraemia, and by the lack of ultrasound findings consistent with urinary disease and negative urine cultures. They can also be differentiated from the condition described by Spitzer-Weinstein, characterised by a pattern similar to Gordon's syndrome but without hypertension, due to the younger age of presentation, the need to combine sodium bicarbonate with thiazides to normalise the internal environment, and the transient nature of the condition.18
Two patients had a marked decrease in FEK+ (1.9% and 3.2%); the patient in whom the transtubular potassium gradient was calculated consistently had a low value compatible with decreased FEK+. In addition, it is worth mentioning that in the 45-day-old patient, despite having a normal value of 13.2%, the FEK+ was also considered inappropriate for his hyperkalaemia, as in situations of acute and chronic hyperkalaemia, FEK+ should increase dramatically.19
As we mentioned earlier, the pathophysiology of the patients reported by McSherry was attributed to a probable maturational disorder of the mineralocorticoid receptors of the distal tubule.1,2 However, this theory does not explain the absence of salt wasting that would be expected along with K+ retention because aldosterone cannot adequately bind to its tubular receptor, as occurs in pseudohypoaldosteronism type 1 or secondary to urinary infection and/or urinary tract disease.3,17
Studies carried out in newborn mammals have observed an adequate number of receptors for aldosterone and of binding sites for the hormone-receptor complex at the nuclear level, so it is now thought that early hyposensitivity to aldosterone is a post-receptor phenomenon.19 This could explain the dissociation from the normal response to aldosterone observed in our patients, who abnormally retained K+ but continued to have adequate reabsorption of Na+. In fact, in an early phase, once in the nucleus, the mineralocorticoid-receptor complex causes the activation and repression of genes capable of modulating the activity of the main Na+ and K+ transporters already in the distal tubular segments, especially the Na+/K+ ATPase pump, the thiazide-sensitive Na+/Cl– co-transporter (NCC) and the epithelial sodium channel (ENaC).20 Among these early aldosterone-induced genes are also different kinases, including WNK4 (lysine-deficient kinases), Sgk-1 (serum- and glucocorticoid-regulated kinase 1), and those of the Src family of protein tyrosine kinases (SFKs).20–24 Subsequently, in its late phase, aldosterone directly modulates the levels of expression of the different transporters of Na+ and K+,20 and so contributes to the reabsorption of Na+ by stimulating the Na+/K+ ATPase pumps, the NCC and the ENaC; and also indirectly through intervention of Sgk-1, which stimulates ENaC and Na+/K+ ATPase pump activity.20,22 In relation to the management of K+, the action of aldosterone is produced by different kinases modulating the actions of the renal outer medullary potassium (ROMK) channels, which regulate cation efflux in the collecting tubule.20,21 WNK4 inhibits expression of ROMK channels, reducing the secretion of K+, an action which is then reversed by Sgk1.21,23,25 In addition, the inhibition of WNK4 by SgK1 is attenuated by the SFK in order to prevent K+ secretion in the absence of hyperkalaemia.23 This attenuation is also observed in hypovolaemia, in which there is an increase in aldosterone and SgK1 activity, leading to Na+ retention and increased K+ secretion. However, if concomitantly K+ influx is low, as previously mentioned, SFKs attenuate SgK1, whereby WNK4 restores ROMK inhibition, decreasing K+ secretion. In this situation we can see how the effects of aldosterone can become dissociated.23 Another situation where this dissociation is observed is in patients with Gordon's syndrome (pseudohypoaldosteronism type 2), in which inhibitory mechanisms other than mutated WNK4 on NCC and ROMK channels could explain the uncoupling. Nonsense mutations of these kinases cause loss of inhibition of NCC expression, generating Na+ retention and hypertension on the one hand and, on the other, increased inhibition of ROMK channels, which leads to hyperkalaemia.26 Similarly, the lack of coordination between the complex mechanism of regulation and counter-regulation of kinases, especially WNK4 as a multifunctional regulator that can dissociate the effects of aldosterone on Na+ and K+ secretion, could be responsible for what we observed in our patients, who reabsorbed Na+ normally but had decreased K+ secretion. This could represent the accentuation of a necessary physiological process in newborns and infants in the first few months, as they need a positive balance of K+ and Na+ to cope with the rapid somatic growth they undergo. This is essentially achieved thanks to retention of these electrolytes by the CCD, as a result of which they also have higher serum K+ levels than older infants and children.25,27,28 Furthermore, as shown in animal models, in the first three weeks of postnatal life there is a lack of expression of the ROMK channel protein from the principal cells of the CCD, with K+ excretion only reaching levels comparable to those of an adult at six weeks of life.29 Along the same lines, the Maxi K+ channel, which is the other channel involved in K+ secretion in CCD, particularly in response to increased distal tubular flow, is only expressed after four weeks of postnatal life,29,30 so it could be another contributing factor to hyperkalaemia. These observations would also explain the fact that it is a transient tubular disorder, as occurred in our patients.
In conclusion, the evidence suggests that TECH without salt wasting may be the exaggerated expression of a maturational phenomenon, characterised by a later expression of the channels that regulate K+ balance in the CCD under the influence of aldosterone, whose maturation is normally slow, in order to ensure the high K+ retention necessary for rapid somatic growth. As it is a transient condition with few clinical manifestations, it should be considered among the causes of hyperkalaemia and growth retardation in the first months of life.
Conflicts of interestNothing to declare.
This study received no specific funding from public, private or non-profit organisations.
This study was approved by the ethics committee at our institution.
Please cite this article as: Alvarado C, Balestracci A, Toledo I, Martin SM, Beaudoin L, Voyer LE. Hiperpotasemia transitoria del lactante sin pérdida salina, enfoque fisiopatológico de tres casos. Nefrologia. 2022;42:203–208.



