


Knowledge of the signalling pathways involved in various diseases has enabled advances in the understanding of pathophysiological, diagnostic and therapeutic models of several inflammatory and autoimmune diseases. Systemic lupus erythematosus is a widely studied autoimmune disease that can affect multiple organs, with a major impact on morbidity and mortality when it involves the kidneys. Over the past 10 years, interest in the role of the TWEAK/Fn14 signalling pathway in lupus nephritis, as well as other clinical settings, has increased. By reviewing the literature, this article assesses the role of this pathway in lupus nephritis, underlines the importance of TWEAK in urine (uTWEAK) as a biomarker of the disease and stresses the favourable results published in the literature from the inhibition of the TWEAK/Fn14 pathway as a therapeutic target in experimental animal models, demonstrating its potential application in other settings. Results of ongoing clinical trials and future research will give us a better understanding of the real benefit of blocking this pathway in the clinical course of several conditions.
El conocimiento de las vías de señalización implicadas en distintas enfermedades ha permitido avances en el entendimiento del modelo fisiopatológico, diagnóstico y terapéutico de varias enfermedades inflamatorias y autoinmunes. El lupus eritematoso sistémico es una enfermedad autoinmune ampliamente estudiada, la cual puede afectar múltiples órganos, con un importante impacto en la morbimortalidad cuando existe afectación renal. Durante los últimos 10 años ha aumentado el interés sobre el papel de la vía de señalización del TWEAK/Fn14 en la nefritis lúpica al igual que en otros escenarios clínicos. Este artículo realiza una revisión de la literatura del papel de esta vía dentro de la nefritis lúpica, recalca la importancia del TWEAK en orina (uTWEAK) como biomarcador de la enfermedad, indica los resultados favorables obtenidos en la inhibición de la vía del TWEAK/Fn14 como diana terapéutica en modelos experimentales animales publicados en la literatura y muestra su posible utilidad en otros escenarios. Los diferentes ensayos clínicos en curso y otras futuras investigaciones darán un mejor panorama en cuanto al beneficio real del bloqueo de esta vía en el curso clínico de estas enfermedades.
Systemic lupus erythematosus (SLE) is an autoimmune disease of multifactorial aetiology, characterised by the formation of immune complexes (antigen–antibody) that are deposited in different organs, activation of the complement system, infiltration of inflammatory cells in target tissues, lymphocyte hyperreactivity and loss in clearance of immune complexes by the reticuloendothelial system.1 The change in homeostasis in the immune system, is the cause of tissue damage and the secondary systemic involvement. Renal impairment due to SLE occurs in about 50% of patients at some point of the disease and it is an indicator of morbidity and mortality.2 Lupus nephritis (LN) is characterised by a diversity of clinical presentation. Glomerular involvement causes haematuria, proteinuria and the progressive deterioration of renal function. The histological pattern is dynamic and changing, so its understanding is key for treatment and prognosis.
Despite advances in knowledge of the disease's pathophysiology and the currently available therapeutic arsenal, the response rates to therapy (complete remission+partial remission) in LN do not exceed 60%.2 Around 25% of patients with renal impairment due to SLE will develop glomerulosclerosis, which requires renal replacement therapy.
The recent knowledge of the TNF-related weak inducer of apoptosis (TWEAK/Fn14) signalling pathway has helped to establish its role in the pathophysiology of LN's. TWEAK is a type II membrane glycoprotein that belongs to the TNF superfamily, and it is produced endogenously in low concentrations by immune cells (monocytes/macrophages) and non-immune cells in different tissues, for example, renal cells, endothelial cells, synovial membrane, intestinal mucosa. Fn14 is a type I membrane protein from the TNF receptor superfamily, and its specific ligand is TWEAK.3 Experiments performed first in animals and then in humans have demonstrated the role of TWEAK/Fn14 in LN.4 Although the TWEAK/Fn14 pathway is not specific to this condition, its understanding will enable the development of future medical interventions that may modify the natural course of the disease.
This paper discusses the pathophysiological model of the TWEAK/Fn14 pathway in LN; it stresses the importance of urinary TWEAK (uTWEAK) as a biomarker of the disease; indicates the favourable results obtained by the inhibition of the TWEAK/Fn14 pathway as a therapeutic target in animals experiments; and defines the basis for future research.
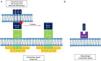
Physiological role of TWEAK/Fn14Molecular structureTWEAK is a 249-amino-acid (aa) type II membrane glycoprotein expressed by immune cells (monocytes/macrophages) and non-immune cells, and the endogenous physiological production is low. Its C-terminal extracellular domain is susceptible to proteolysis by proteases that allow the soluble form of the molecule (sTWEAK) to circulate freely. The N-terminal intracellular domain also possesses regions susceptible to cleavage by proteases that facilitate the exposure of nuclear localisation sequences, the function of which is so far uncertain.5 Because of its structural homology with TNF, its biological activity depends on the formation of trimers for specific binding to the Fn14 receptor (CD 266). Fn14 is a 129-aa type I membrane protein expressed in low concentrations in cells and it belongs to the TNF receptor superfamily. Its N-terminal extracellular domain features serine-rich regions involved in the binding to its specific ligand, TWEAK. The C-terminal intracellular domain helps to recruit TRAF-like proteins (factors associated with the TNF receptor) that start the intracellular signalling cascade. Unlike other TNF superfamily receptors that recruit FADD, TRADD, and TRAF complexes, Fn14 only recruits TRAF5-like complexes (Fig. 1). This difference is very important because it explains the weak apoptotic capacity of TWEAK, which can only be mediated by the mitochondrial pathway and not by death receptors, particular to the extracellular pathway.
(A) TWEAK/Fn14 interaction. The Fn14 receptor is capable of binding mTWEAK and sTWEAK and helping to recruit TRAF-like domains, which start the intracellular signalling cascade. (B) The macrophage scavenger receptor type is also able to bind sTWEAK and facilitate its internalisation in the cytoplasm. NLS: nuclear localisation signal; sTWEAK: soluble TWEAK.
Likewise, it has been possible to demonstrate how the only ligand for Fn14 is TWEAK. The scavenger receptor type CD163 expressed by macrophages has the ability to bind TWEAK and facilitate the internalisation and subsequent depuration of the molecule's soluble form (sTWEAK); it is therefore thought that this mechanism constitutes a negative regulation pathway of TWEAK/Fn14.6
Signalling effector pathwaysOnce mTWEAK or sTWEAK binds to its Fn14 receptor, the intracellular signalling pathway may result in one of 3 effector pathways: inflammatory, proliferative or apoptotic (Fig. 2). Perhaps the best characterised pathway is the inflammatory one, since it is similar cytoplasmic signalling pathways in other cell populations. This effector pathway depends on the canonical or non-canonical activation of the transcription factor of NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells). The mTWEAK/Fn14 interaction promotes the predominance of canonical activation signals of NF-kB with the formation of the RelA/p50 heterodimer in the effector cell cytoplasm. By contrast, the sTWEAK/Fn14 interaction promotes the non-canonical activation signals of NF-kB via the RelB/p52 heterodimer.5,6 From a clinical point of view, this difference in the way NF-kB is activated might not be of major significance. However, from an immunological point of view, differences in the type of genes transcribed and the type of cells in the immune system recruited into the target tissue are fundamental. The early canonical activation of NF-kB (RelA/p50) promotes the synthesis and secretion of monocyte chemoattractant protein-1 (MCP-1), chemokine CCL5 (RANTES) and chemokine CXCL10 (IP-10). This gradient of chemokines allows the tissue infiltration of macrophages. By contrast, the late non-canonical activation of NF-kB (RelB/p52) promotes the synthesis of chemokines CCL19 and CCL21, the gradient of which allows T lymphocytes to infiltrate the target organs.
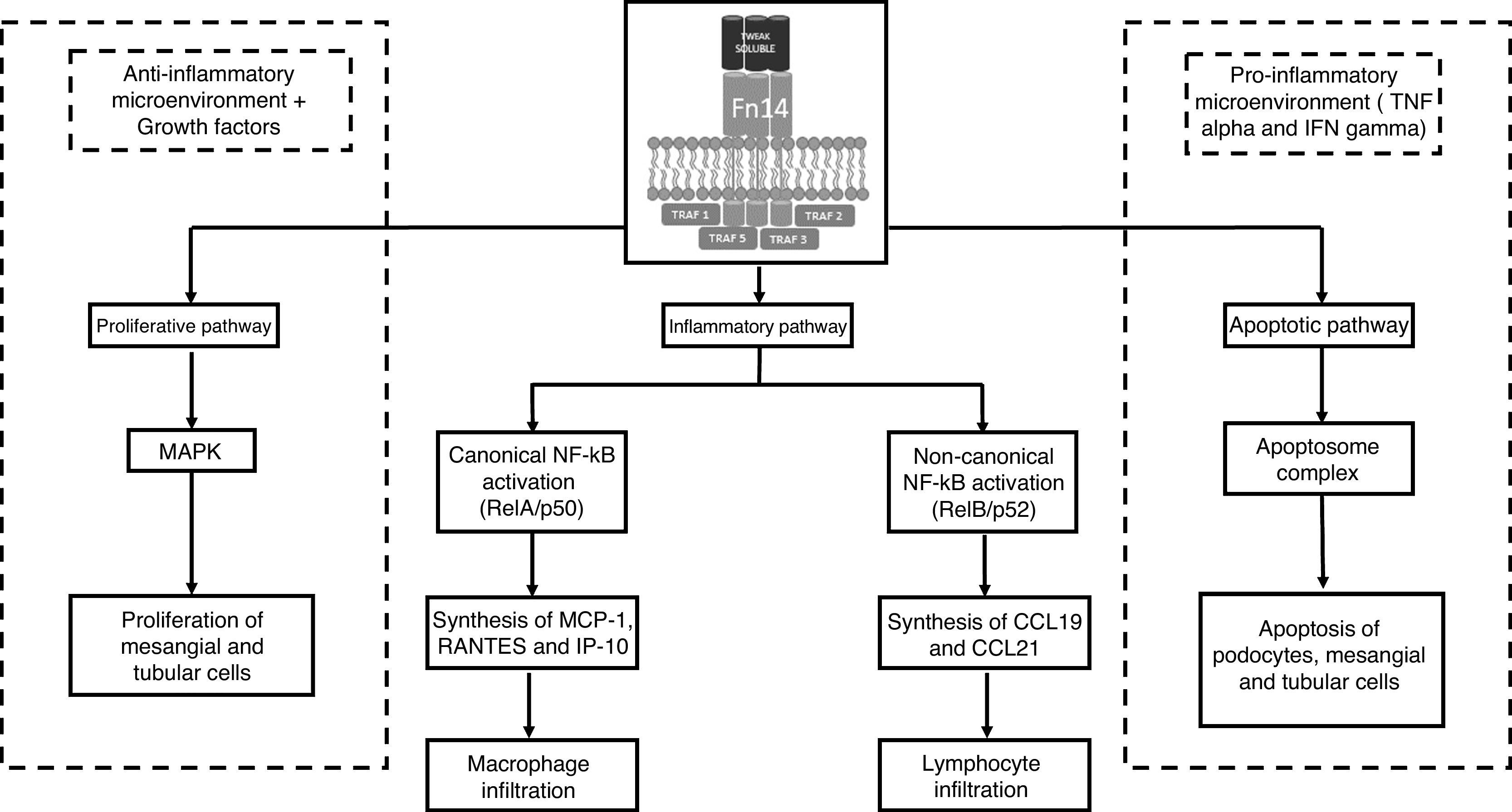
Role of TWEAK/Fn14 in lupus nephritis. The binding of TWEAK to the Fn14 receptor encourages the activation of one of the 3 effector pathways, depending on the type of cell stimulated, the cellular microenvironment and the intracellular signalling generated. IP-10: interferon-gamma-inducible protein 10; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemoattractant protein type 1; NF-kB: nuclear factor kappa B.
The activation of the 2 effector pathways remaining as a result of the TWEAK/Fn14 interaction will depend specifically on the existing cellular microenvironment. In a cellular environment where there is no inflammation and which is rich in growth factors, the dominant effector pathway will be the proliferative one. By contrast, in the presence of proinflammatory cytokines (TNF α-IFN γ), the apoptotic effector pathway will be the dominant one. The opposite effect in these 2 effector pathways lies mainly in the different cytoplasmic concentrations of antiapoptotic (Bcl2) and proapoptotic (Bax) protein complexes, respectively.7 In the proliferative effector pathway there is an increase in mitogen-activated protein kinase (MAPK) activity, whereas in the apoptotic effector pathway there is a functional increase of the apoptosome complex (APAF-1, cytochrome C, procaspase-9) that mediates programmed cell death via the mitochondrial pathway.8
These findings in the proliferative effector pathway have been demonstrated by Sanz et al., in compensatory renal hyperplasia after a unilateral nephrectomy, where there is evidence of TWEAK/Fn14-induced renal tubular epithelium proliferation.9 Also, according to Gao et al., the stimulation of TWEAK/Fn14 contributes to the proliferation of human renal cells (mesangial, podocyte and tubular epithelial cells) in in vitro and in vivo models.10
Role of TWEAK/Fn14 in lupus nephritisAs described above, in states of cellular rest, without stimuli associated with injury and inflammatory, the tissues’ expression of TWEAK and Fn14 is low.11 However, in inflammatory events such as SLE, there is an increase in Fn14 receptor expression in renal cells, a rise that is not proportional to mTWEAK/sTWEAK availability despite these proinflammatory conditions.4 In addition, it has been possible to establish, e.g. in patients with LN, an increase in Fn14 expression in 3 types of renal cells: podocytes, glomerular mesangial cells and the proximal tubular epithelium. This phenomenon may in part explain the mesangial proliferation, podocytopathy and tubular damage observed in the biopsies of patients with renal impairment due to SLE. The activation of the above-mentioned inflammatory pathway contributes to the glomerular and tubular infiltration of macrophages; the direct cytotoxicity mediated by these cells; and the mesangial and tubular apoptosis – all events that progressively deteriorate renal function. All these changes added to the activation of the complement system and the deposit of immune complexes accelerate kidney injury.
In current clinical practice, markers of disease activity have limitations, given their low sensitivity and specificity. From an immunological point of view, only 2 markers are used on a regular basis to track activity in LN: serum complement levels (C3–C4) and anti-dsDNA antibody titres.12,13 This is why research in recent years has focused on the search for new biomarkers able to predict in advance a relapse of the disease so, a more aggressive immunosuppressive regimen is applied and renal damage is prevented.14 Urine, as a biological sample from glomerular filtration, may directly reflect the inflammatory and cytotoxic events occurring during a relapse. Proteomic studies in urine samples in patients with SLE with renal impairment have made possible to recognise potential biomarkers that could serve as predictors of disease activity.15 By using a proteomic analysis of urine, Mosley et al. were able to identify 2 proteins as candidate biomarkers for distinguishing between active and inactive LN, and for differentiating between disease relapse and remission.16
Xuejing et al. found that after analysing urine samples from 46 SLE patients, with or without secondary renal impairment, uTWEAK levels were significantly higher in those with active LN vs. those without active LN.17 These uTWEAK levels correlated directly with the index of total activity and the index of glomerular and tubulointerstitial activity, but not with the index of chronicity. Moreover, there was a significant relationship between uMCP-1 levels and renal activity due to SLE.
Other studies, such as the one by Schwartz et al. in 2006 in which 2 cohorts of SLE patients were studied; the Ohio SLE Study (OSS); and the Albert Einstein College of Medicine (AECOM) study, showed a direct correlation between uTWEAK levels and rSLEDAI, a rating system used to assess the degree of renal activity in patients with SLE.18 Likewise, and as expected, there was a correlation directly proportional between uTWEAK levels and anti-dsDNA titres, and inversely proportional to serum complement (C3–C4) levels. Interestingly, the type of immunosuppressive therapy used did not have a significant influence in the change in uTWEAK levels; this suggest the therapeutic arsenal currently available does not block the TWEAK/Fn14 signalling pathway. This same group19 showed that there was a statistically significant difference between uTWEAK levels, not only between patients with active and inactive LN, but also with respect to healthy individuals and patients with rheumatoid arthritis (RA) and osteoarthritis. Thus, despite the fact that there is an inflammatory state in these other clinical scenarios, uTWEAK may be a more specific biomarker of renal involvement in SLE.
Other urine biomarkers in SLENot only has uTWEAK been identified as a potential biomarker in LN, but also, in 2011, El-Shehaby et al. showed how levels of uMCP-1, urine osteoprotegerin (uOPG) and urine IL-8 were significantly higher in patients with active LN than in those without LN.20 There was also a direct correlation between uTWEAK, uMCP-1, urine osteoprotegerin levels and the rSLEDAI score.
Hutcheson et al.21 evaluated the role of adipokines, proteins derived from adipose tissue, in both blood and urine, and their usefulness as biomarkers for predicting renal impairment due to SLE in 38 LN patients. The authors found that patients with LN had higher serum levels of adiponectin, leptin and resistin as compared with healthy control groups. However, urine resistin was the only one that showed predictive capacity. Of all the adipokines evaluated, only serum resistin was directly correlated with indicators of kidney injury (creatinine, BUN and proteinuria/creatininuria index).
Much research has been done to identify the usefulness of these biomarkers for predicting not only the clinical course of the disease but also the type of histology of renal involvement in SLE,22–24 which plays a fundamental part in the diagnosis, treatment and prognosis of the disease. Studies such as the one by Brunner et al.,25 for example, identified how the combination of uMCP-1, urinary alpha-1 acid glycoprotein (uAAG) and urine ceruloplasmin were excellent biomarkers for predicting active proliferative LN when added to the use of the proteinuria/creatininuria index as a clinical marker of the disease. They also found that urinary neutrophil gelatinase-associated lipocalin (uNGAL) and uMCP-1 in combination with serum creatinine clearance as a clinical marker were predictors of LN chronicity, and, as with uAAG, C4 serum levels together with creatinine clearance were excellent predictors of membranous LN. Despite these findings, to date there is no biomarker capable of replacing renal biopsy as the gold standard for the diagnosis of the type of LN in progress.
The usefulness of other urine biomarkers in SLE and other glomerulopathies is beyond the scope of this article, but they have been reviewed recently.26,27
TWEAK/Fn14 in other clinical settingsBased on the full characterisation of the TWEAK/Fn14 signalling pathway, it has been possible to investigate in clinical settings other than the model of renal impairment due to SLE. Without question, one of the fields in which the most progress has been made is that of atherosclerotic cardiovascular disease. Like diabetes mellitus, chronic kidney disease (CKD) is considered a cardiovascular risk factor. In a cohort of 1058 patients with stages 3-5D CKD and no known history of cardiovascular disease, Fernández-Laso et al.28 found that serum levels of sTWEAK decreased as the stage of CKD progressed. They also found that those individuals with a drop in sTWEAK levels had a higher degree of thickening of the carotid intima-media, a trend that was maintained with respect to the presence and severity of atherosclerotic plaques – calcified or not – on this level. Once the variables were adjusted for the Cox logistic regression model, it was found in this cohort of patients that individuals with lower serum levels of sTWEAK had a higher rate of fatal and non-fatal cardiovascular events. This drop in serum concentrations of sTWEAK, relative to the severity of the endothelial injury, seems to be explained by the increased binding of sTWEAK to Fn14, expressed by the injured endothelial cells, which significantly decreases the serum levels of sTWEAK.
In another study, Akdoğan et al.29 evaluated the serum levels of sTWEAK and MCP-1 in a cohort of 97 patients with stages 2–3 CKD, who underwent heart catheterisation for invasive stratification of coronary disease using the Gensini score (the maximum score of 32 points indicates 100% coronary occlusion). It is striking that they found a directly proportional relationship between the levels of sTWEAK and MCP-1 and the Gensini score obtained.
Jasiewicz et al.30 evaluated the relationship of TWEAK in other cardiovascular scenarios. This group performed a comparative study of 26 patients with pulmonary hypertension, confirmed by right heart catheterisation and 24 healthy volunteers. Their results showed that individuals with pulmonary hypertension had a higher expression of the CD163 marker (macrophage scavenger receptor) and lower serum levels of sTWEAK as compared with healthy volunteers. The explanation for these results is not entirely clear, but as previously mentioned, the fact that sTWEAK binds to Fn14 might explain the reduction of serum concentration. Also, the increased expression of the CD163 receptor might indicate an activation of the negative regulation mechanisms of TWEAK/Fn14. These are, however, unconfirmed hypotheses.
Given the structural homology between TNF and TWEAK, TNF's leading role has been extrapolated to the pathophysiology of other diseases and the relation to the TWEAK signalling pathway. Conditions like inflammatory bowel disease (IBD), psoriasis and RA are characterised by high levels of TNF alpha in affected tissues. For example, the expression of TWEAK messenger RNA in the mucosa of patients with IBD has been described with an exponential and direct increase according to the disease's activity.31 Furthermore, Bilgiç et al.32 studied the differences in the expression of TWEAK and other cytokines (IL-6, IL-23, TNF alpha) in serum in 45 patients with chronic psoriasis and 43 healthy volunteers. Their results showed that serum sTWEAK, IL-6, IL-23 and TNF alpha were significantly higher in individuals with chronic psoriasis than in healthy volunteers, but these findings did not correlate with disease severity as estimated using the Psoriasis Area and Severity Index (PASI). In RA, an increase in sTWEAK concentrations in the synovial fluid of patients with active disease has been found, in addition to an increase in RANKL expression by sTWEAK-induced synoviocytes33 and osteoblasts.34 Compared with psoriatic arthritis, patients with active RA have higher levels of sTWEAK, which in part explains the potential joint damage this disease can cause.35
Finally, sTWEAK has been used as a biomarker in terms of recovery in endothelial function following kidney transplantation. In 2013, Yilmaz et al.36 published a study in which they followed a cohort of 175 patients with kidney transplantation for 6 months. They evaluated endothelial function by measuring flow-mediated dilation of the brachial artery with Doppler ultrasound on day 0 and 180 days post-transplantation. They likewise evaluated the change in serum concentrations of sTWEAK over the same time period. Their results showed that improvement in Doppler blood flow correlated directly with normalisation in serum concentrations of sTWEAK, possibly in relation to its lower degree of binding to the Fn14 expressed by endothelial cells.
TWEAK/Fn14 as a therapeutic targetSeveral studies in experimental models in animals and also human studies have evaluated the usefulness of blocking the TWEAK/Fn14 signalling pathway37 (Table 1). Studies performed on animals with acute kidney injury following folic acid overdose have shown that blocking TWEAK/Fn14 is associated with improved renal function, reduced interstitial inflammation, decreased proliferation and tubular death.38 These findings were also reproduced in another animal experiment with acute kidney injury through the induction of kidney damage caused by ischaemia-reperfusion injury.39 In this work, for example, a decrease was also shown in residual renal interstitial fibrosis when TWEAK/Fn14 was blocked.
Murine experimental models of the TWEAK/Fn14 pathway as a therapeutic target in various clinical scenarios.
| Author (year) | Clinical scenario | Methodology | Results |
|---|---|---|---|
| Desplat-Jego et al. (2005)45 | Experimental autoimmune encephalomyelitis | Anti-TWEAK | Decreased severity score (40–80%) and leucocyte infiltration in brain tissue |
| Perper et al. (2006)46 | Collagen-induced arthritis | Anti-TWEAK | Reduced arthritogenic mediators in serum levels (RANTES, IP-10, MCP-1, CCL4, CXCL1) |
| Zhao et al. (2007)47 | Nephritis induced by graft-versus-host disease (SLE) | Anti-TWEAK | Decreased expression of IL-6, MCP-1 and IL-10 by kidneys and reduced proteinuria |
| Sanz et al. (2008)38 | Acute kidney injury from folic acid overdose | Anti-TWEAK | Decreased expression of MCP-1 and RANTES by 60% in renal tubular epithelium |
| Sanz et al. (2009)9 | Acute kidney injury after unilateral nephrectomy (non-inflammatory model) | TWEAK−/− (knockout) mice | Reduced tubular proliferation and apoptosis, improved renal function |
| Muñoz-García et al. (2009)48 | Nephropathy induced by hyperlipidaemic diet | ApoE−/− (knockout)+Anti-TWEAK mice | Decreased expression of RANTES and MCP-1, macrophage infiltration and severity of kidney injury |
| Hotta et al. (2011)39 | Acute kidney injury due to ischaemia-reperfusion injury | Anti-Fn14 | Reduced inflammatory cytokines, macrophage-neutrophil infiltration at the tubular level |
| Xia et al. (2012)40 | Nephrotoxic serum nephritis | Fn14+/+ WT (wild type) vs. Fn14−/− KO (knockout) mice | Reduced proteinuria, tubular disease and glomerular crescent formation |
Xia et al. reproduced in a model of nephrotoxic nephritis, the immunological basis that started from the deposit of immune complexes at the glomerular level, thus simulating the conditions of LN, to demonstrate the clinical, paraclinical and histopathological course in a murine model by blocking the TWEAK/Fn14 pathway.40 Fn14+/+ WT (wild type) and Fn14−/− KO (knockout) mice were analysed comparatively. The Fn14−/− KO mice had a lower degree of proteinuria, less tubular disease and less glomerular crescent formation as compared with the Fn14-expressing strains. The use of anti-TWEAK monoclonal antibody injected in mice that did not die from uraemia after being inoculated with nephrotoxic serum showed a lower intensity in the Periodic acid–Schiff staining, a lower proliferation index (Ki 67+) and less glomerular infiltration by macrophages in each of the biopsies analysed.
Despite all of these potentially beneficial effects of blocking the TWEAK/Fn14 pathway in different diseases, not all studies have shown favourable results. In a murine model, Mustafa et al. explored whether the genetic or pharmacological ablation of TWEAK/Fn14 modified the histological changes of the substantia nigra and striated in Parkinson's disease.41 Their results failed to demonstrate a reduction in acute neurotoxicity mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine after the genetic ablation of TWEAK and Fn14.
There are phase I and phase II studies of humans in progress that assess the role of TWEAK/Fn14 as a therapeutic target in RA,42 advanced solid tumours43 and LN.44 Their results will provide a better picture of the usefulness of blocking this pathway in these clinical scenarios.
Conclusions and future perspectivesSignificant progress has been made in casting light on the TWEAK/Fn14 pathway in the pathophysiological model of several types of autoimmune and inflammatory diseases. For the time being, it is clear that TWEAK/Fn14 is involved in the processes of inflammation, proliferation and apoptosis of podocytes, glomerular mesangial cells and tubular epithelial cells of patients with renal impairment due to SLE. Based on the current evidence, we might consider the TWEAK/Fn14 pathway to be a necessary but insufficient mechanism to fully explain the potentially harmful events caused by this type of disease. The clinical usefulness of blocking the TWEAK/Fn14 pathway being evaluated in studies in progress will determine the real importance of this molecule as the leading actor in LN and other clinical scenarios.
- -
The activation of the TWEAK/Fn14 pathway in LN contributes to the infiltration of macrophages and lymphocytes on the glomerular and tubular levels; the direct cytotoxicity mediated by these cells; and mesangial and tubular apoptosis.
- -
The TWEAK/Fn14 signalling pathway participates in the pathogenesis of a number of chronic inflammatory processes, such as psoriasis, inflammatory bowel disease, rheumatoid arthritis and end-stage chronic kidney disease, among others.
- -
Urinary TWEAK levels have been related to active LN and have been directly correlated with the total activity index and the glomerular and tubulointerstitial activity index in kidney biopsies.
- -
Blocking the TWEAK/Fn14 pathway is an attractive therapeutic target in LN and other chronic inflammatory diseases with various ongoing phase I and phase II studies.
The authors declare that they have no conflicts of interest.
José A. Gómez-Puerta received support from “Colciencias” (call 656 of 2014).
Please cite this article as: González-Sánchez DA, Álvarez CM, Vásquez G, Gómez-Puerta JA. Papel de la vía de señalización del TWEAK/Fn14 en la nefritis lúpica y otros escenarios clínicos. Nefrologia.2017;37:118–125.



