
To demonstrate that the variant not described in PKD1 gene c.7292T> A, identified in four families from the Alpujarra in Granada, is the cause of autosomal dominant polycystic kidney disease (ADPKD). This variant consists of a transversion of thymine (T) by adenine (A) that at the level of the Polycystin 1 protein produces a change of leucine (Leu/L) by Glutamine (Gln/Q) in position 2431 (p.Leu2431Gln).
MethodSociodemographic and clinical variables were registered using clinical histories, genealogical trees, ultrasounds and genetic analysis to ADPKD and healthy individuals belonging to these families in the context of segregation study.
ResultsAll PKD individuals carried the c.7292T> A variant in heterozygosis, whereas healthy ones did not. Among all ADPKD patients, 62.9% were women. ADPKD diagnosis was made at 29.3 ± 15.82 years, after having the first child in 64.8%. The main reasons for diagnosis were family history and hematuria episodes. The onset of renal replacement therapy (RRT) occurred at 55.8 ± 7.62 years (range 44–67), and death at 63 ± 92.2 years (range 48–76), being the cause unknown, cardiovascular and insufficiency kidney the most frequent; the median of renal survival was established at 58.5 ± 0.77 years and the median survival of patients at 67.2 ± 3.54 years. No differences in kidney and patient survivals were observed according to sex. Among deceased patients, 52.2% required RRT and 94.4% suffered from renal failure.
ConclusionsThe variant c.7292T> A in PKD1 gene is responsible for the disease, and its distribution in the Alpujarra region of Granada suggests a founder effect. In ADPKD it is necessary to perform segregation studies that help us to reclassify genetic variants, in this case from indeterminate to pathogenic.
Demostrar que la variante no descrita en el gen PKD1 c.7292T> A, identificada en cuatro familias de la comarca de la Alpujarra de Granada, es la causante de poliquistosis renal autosómica dominante (PQRAD). Esta variante consiste en una sustitución transversión de timina (T) por adenina (A) que a nivel de la proteína Policistina 1 produce un cambio de leucina (Leu/L) por Glutamina (Gln/Q) en posición 2431 (p.Leu2431Gln).
MétodoRegistramos variables sociodemográficas y clínicas a través de la realización de historias clínicas, árboles genealógicos, ecografías y estudios genéticos a individuos afectos y sanos pertenecientes a estas familias en el contexto del estudio de segregación.
ResultadosTodos los individuos afectados portaban en heterocigosis la variante c.7292T> A, mientras que los individuos sanos no la portaron. En las familias estudiadas, el 62.9% eran mujeres. El diagnóstico de PQRAD se realizó a los 29.3 ± 15.82 años de edad, después de haber tenido el primer hijo en el 64,8%. Los motivos principales de diagnóstico de la enfermedad fueron antecedentes familiares y episodios de hematuria. El inicio de tratamiento renal sustitutivo (TRS) se produjo a la edad de 55.8 ± 7.62 años (rango 44–67), y el éxitus a los 63 ± 92.2 años (rango 48–76), siendo la causa desconocida, cardiovascular e insuficiencia renal las más frecuentes; la mediana de supervivencia renal se estableció a los 58.5 ± 0.77 años y la mediana de supervivencia del paciente a los 67 ± 3.54 años. No observamos diferencias en la supervivencia del riñón y del paciente según sexo. De los pacientes fallecidos, el 52.2% necesitaron TRS y el 94.4% tenían algún grado de insuficiencia renal (IR).
ConclusionesLa variante c.7292T> A en el gen PKD1 es responsable de la enfermedad, y su distribución en la comarca de la Alpujarra de Granada sugiere un efecto fundador. En PQRAD es necesario realizar estudios de segregación que ayuden a reclasificar variantes genéticas, en este caso de indeterminada a patogénica.
The polycystic kidney disease autosomal dominant (ADPKD), international codes classified 753.12, 753.13 (ICD-9) and Q61.2, Q61.3 (ICD-10), is the most common hereditary nephropathy causing kidney failure and need for treatment renal replacement therapy (RRT). There are renal cysts that grow progressively replacing the entire normal renal parenchyma and this is accompanied by fibrosis and inflammation of the interstitium.1 It is a monogenic and/or bigenic disease2 that can affect other organs besides the kidney and cause high blood pressure, urinary tract infections, lumbar or abdominal pain, hematuria, nephrolithiasis and kidney failure, as well as cysts of the liver, pancreas and epididymis, choroidal and arachnoid plexus, brain aneurysms, heart abnormalities and abdominal hernias, among others.3 The intensity of these manifestations is variable even between individuals of the same family.4
The prevalence of the disease is not well determined since there are not specific official records, although recent studies indicate that the prevalence is between 4.76–9.14 cases/10,000 births.5 Our registry, made up of 298 families and 1,164 patients identified in the Granada healthcare setting, identifies between 16–31 new cases/year (during the 2000−2018p eriod), with an average incidence of 2.34 cases/105 people/year,6 consistent with data reported in the study Else - Kroener - Fresenius-ADPKD in southwestern Germany.7
Most cases are due to variants in the PKD1 gene (16p13.3), and to a lesser extent in the PKD2 (4q22.1), GANAB (11q12.3) genes8 and DNAJB11 (3q27.3), with few cases of combined involvement of the PKD1 and PKD2 genes.9 These genes encode the proteins polycystin 1 (PKD1), polycystin 2 (PKD2) and the α subunit of glucosidase II (GANAB), which are very ubiquitous in the body, although they are mainly part of the primary cilium.10 Its alteration will cause a decrease in intracellular calcium with an increase in cAMP, alterations in cell proliferation and polarity, an increase in fluid secretion and an increase in the extracellular matrix, all of which are implicated in the cystogenesis, but the exact mechanism is still unknown.10,11
Our aim is to demonstrate that the new variant c.7292T> A identified in the PKD1 gene is responsible for ADPKD in 4 families from the Alpujarra de Granada, apparently not related by a genealogical tree, as well as to identify the existence of a possible founder effect.
MethodsMedical histories, genealogical trees and ultrasounds have been obtained in members of the 4 families affected by PKRAD from the geographical region of the Alpujarra of Granada, that has a registered population of 25,000 people: one family is from Órgiva, with 5,570 inhabitants and the other three from Pórtugos, with 403 inhabitants.12
Genetic studies were performed by massive sequencing (NGS) in people with a diagnosis of ADPKD.13 The variant c.7292T> A (p.Leu2431Gln) was identified in heterozygosity in the PKD1 gene, not described in the databases or in the literature. Additionally the in silico analysis was contradictory: the PoliPhen2, SIFT and Mutation Taster programs indicate a deleterious effect while Align GVGD indicated a benign effect. With this situation it was necessary to plan the segregation study: genetic analyzes were performed in affected relatives (complete sequencing of the PKD1 gene or study of known point mutation, depending on each case) and in healthy relatives (study of known point mutation). In each family, affected and healthy individuals were studied. All participants were informed and signed the consent to participate in the study; in the case of minors parents authorization was requested.
Genetic studies were carried out at the Institute of Genomic Medicine (IMEGEN) using the NextGeneDx ® massive sequencing panel (NGS), which includes the PKD1, PKD2 and GANAB genes, following the following methodology:
- 1
Genomic DNA extraction from the lymphocytes of the blood sample.
- 2
Library preparation using the Nextera XT kit (Ilumina).
- 3
Sequencing of the libraries (2 × 150) in the MiSeq sequencer (Ilumina).
- 4
Bioinformatic analysis of the sequences obtained.
- 5
Confirmation of variants by Sanger sequencing.
Pathogenicity of the variants discovered was evaluated using population databases such as the Human Gene Mutation Database (HGMD), Clinvar, GenomeAD, LOVD and, the one that offers the most specific information, the ADPKD Mutation Database, which identifies 2,601 changes in the structure of the PKD1 gene, between silent, pathogenic and of uncertain or indeterminate clinical significance.14
For point mutations in patients with an already known mutation, Sanger sequencing was performed by the Imegen laboratory.
We recorded sociodemographic, clinical and other variables that could be related to the impact of the mutation on the kidney and the patient by doing survival studies. Geno-Pro programs were used to make the genealogical trees and the statistical package SPSS 15.0. Data are expressed in terms of mean ± SD, range, median, and percentage, as appropriate. Kidney and patient survival studies were performed with the Kaplan-Meier test, considering significance if p < 0.05.
ResultsSociodemographic, clinical and genetic characteristics of the studied population are summarized in Table 1. Due to the accessibility conditions, genetic study was performed in 20 of the 66 patients diagnosed of ADPKD by ultrasound criteria by Ravine and Pei.13 In 8 patients the genetic study was performed by mass sequencing and in 12 by Sanger sequencing to the study of known point mutations. Genetic analysis revealed the presence of the variant c.7292T> A (p.Leu2431Gln) in all affected patients. Likewise, 8 relatives without clinical and ultrasound evidence of the disease were studied and the variant was not identified in any of the 8 relatives.
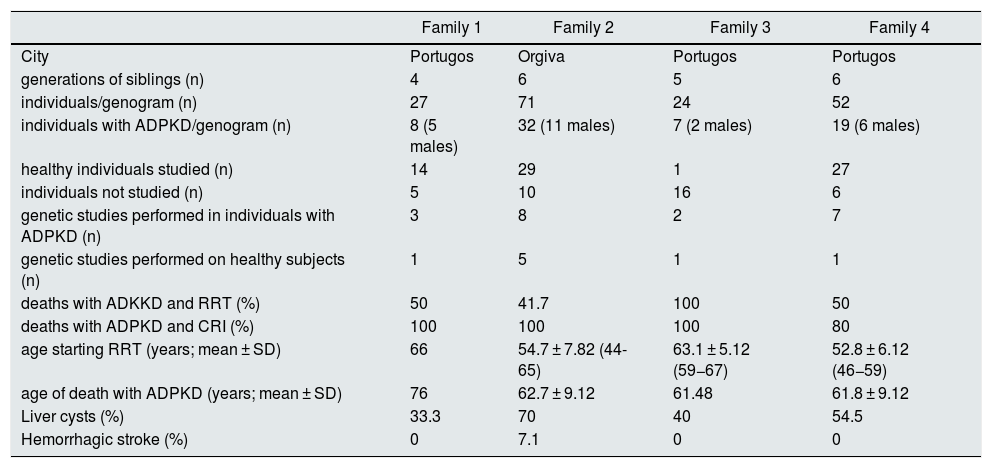
Sociodemographic, clinical and genetic characteristics of the affected families. (CRF: chronic renal failure (eGFR <60 mL/min); RRT: renal replacement therapy).
| Family 1 | Family 2 | Family 3 | Family 4 | |
|---|---|---|---|---|
| City | Portugos | Orgiva | Portugos | Portugos |
| generations of siblings (n) | 4 | 6 | 5 | 6 |
| individuals/genogram (n) | 27 | 71 | 24 | 52 |
| individuals with ADPKD/genogram (n) | 8 (5 males) | 32 (11 males) | 7 (2 males) | 19 (6 males) |
| healthy individuals studied (n) | 14 | 29 | 1 | 27 |
| individuals not studied (n) | 5 | 10 | 16 | 6 |
| genetic studies performed in individuals with ADPKD (n) | 3 | 8 | 2 | 7 |
| genetic studies performed on healthy subjects (n) | 1 | 5 | 1 | 1 |
| deaths with ADKKD and RRT (%) | 50 | 41.7 | 100 | 50 |
| deaths with ADPKD and CRI (%) | 100 | 100 | 100 | 80 |
| age starting RRT (years; mean ± SD) | 66 | 54.7 ± 7.82 (44-65) | 63.1 ± 5.12 (59−67) | 52.8 ± 6.12 (46−59) |
| age of death with ADPKD (years; mean ± SD) | 76 | 62.7 ± 9.12 | 61.48 | 61.8 ± 9.12 |
| Liver cysts (%) | 33.3 | 70 | 40 | 54.5 |
| Hemorrhagic stroke (%) | 0 | 7.1 | 0 | 0 |
In these 4 families, a 62.9% of affected were women. Diagnosis was made at 29.3 ± 15.82 years of age (age range 1–59), in 64.8% after delivery of a first child. The presence of a family history and hematuria were the two main causes that led to the diagnosis. In affected relatives, hepatic cysts were identified in 59.3%, a history of hemorrhagic stroke in 3.3%, and cysts in the epididymis or ovary in 13%. The age of initiation of renal replacement therapy (RRT) was 55.8 ± 7.62 years (age range 44–67); death occurred at 63.1 ± 9.32 years (range 48–76 years), the most frequent causes were: unknown cause, cardiovascular and renal failure frequently. The median renal survival was 58.5 ± 0.77 years and the patient's median survival was 67.2 ± 3.54 years, without significant differences based on sex. Of the deceased patients, 52.2% required RRT and 94.4% had some degree of renal failure (IR).
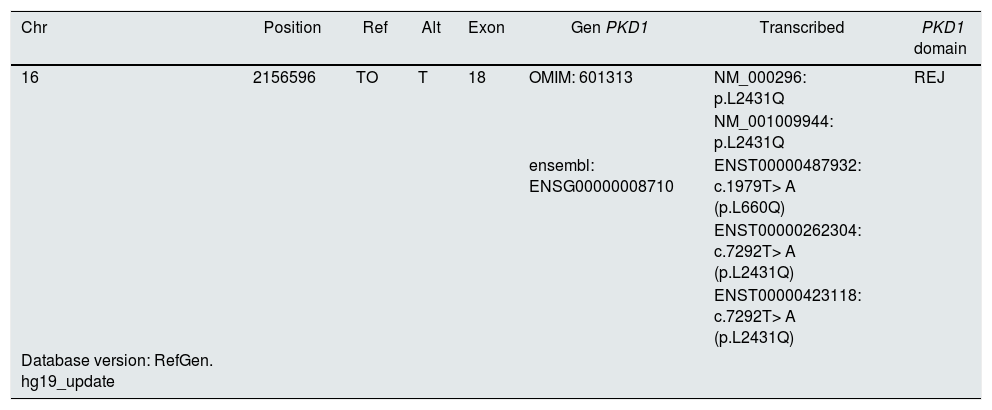
DiscussionOur variant, not identified in the general or specific human gene variant registries, is located in exon 18 at chromosomal position chr16: 2156596 where an adenine to thymine transversion substitution occurs that predicts a missense mutation (missense) with the modification of leucine (L) by glutamine (Q) at codon 2431 (c.7292T> A, L2431Q) (Table 2).
Characterization of the variant according to different transcripts of the PKD1 gene.
| Chr | Position | Ref | Alt | Exon | Gen PKD1 | Transcribed | PKD1 domain |
|---|---|---|---|---|---|---|---|
| 16 | 2156596 | TO | T | 18 | OMIM: 601313 | NM_000296: p.L2431Q | REJ |
| NM_001009944: p.L2431Q | |||||||
| ensembl: ENSG00000008710 | ENST00000487932: c.1979T> A (p.L660Q) | ||||||
| ENST00000262304: c.7292T> A (p.L2431Q) | |||||||
| ENST00000423118: c.7292T> A (p.L2431Q) | |||||||
| Database version: RefGen. hg19_update |
This change is located in the extramembranous region, REJ domain of polycystin 1 (PKD1). The domain in PKD1 is almost 1,000 amino acids in length between exons 15 and 27. It is likely composed of multiple structural domains. There are six fully conserved cysteine residues that can form disulfide bridges.15 The function of this domain is not well established, although it has been related to the regulation of calcium ion flux.16 Some authors suggest that it is an essential region for the biological activity of polycystin-1.17 The REJ domain is located in the vicinity of the GPS domain (G-protein coupled proteolytic site), which is essential for a normal structure and function of the kidney. Some studies have shown that the GPS domain function requires the integrity of the REJ region. In the Mayo Clinic PKD database there are described around 65 missense variants classified as causes of disease in this region.15 Other missense variants classified as probably pathogenic are described in this same region very close to the protein sequence, such as c.7300C> T, p.Arg2434Trp (R2434W),18 c.7301G> A, p.Arg2434Gln (R2434Q)19 and p. Leu2433Arg (L2433R).20
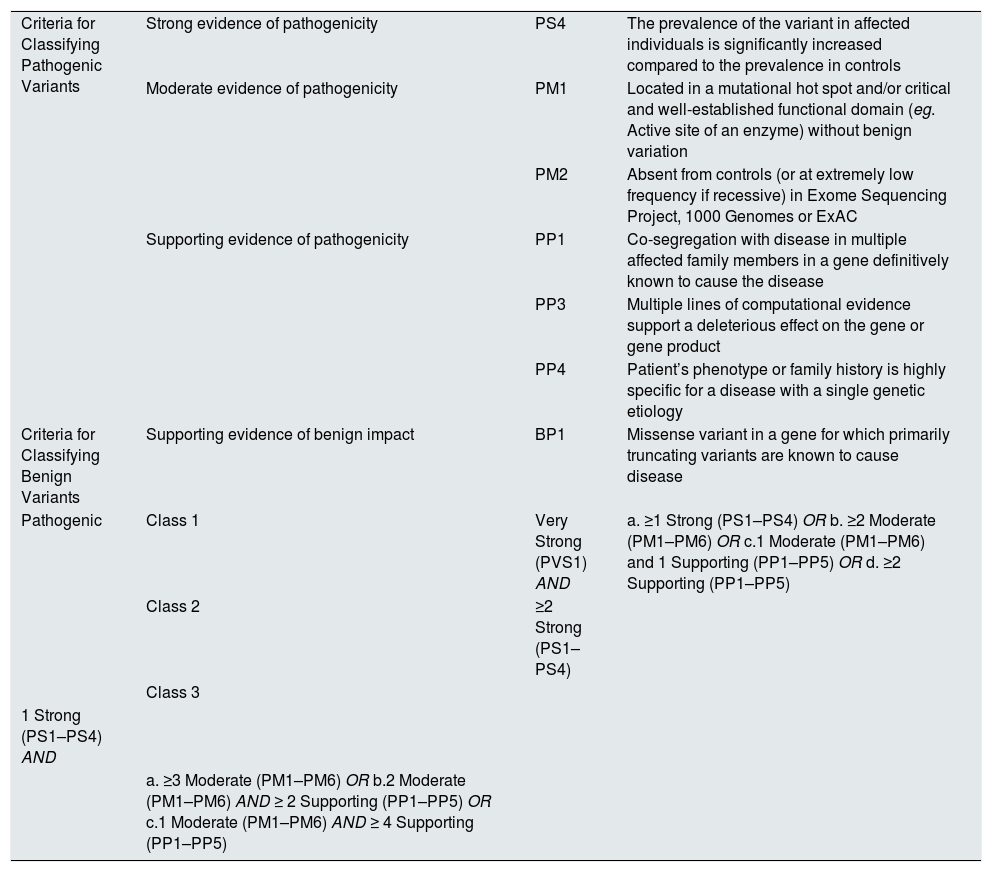
Some in silico predictors classify this variants as deleterious and others as benign. This is not registered in the ExAC, ESP5400 and 1000 Genomes databases which indicates its low frequency in the population. This contradiction of predictors causes the variant to be classified as indeterminate. The segregation study is compelling: the variant is seen in 20 affected people and in none of the 8 healthy individuals studied. That is, the variant set aside with the disease. To classify a variant, it is recommended to follow the criteria of the American College of Medical Genetics and Genomics.21 In 2015, this consensus guide was written trying to actualize, as far as possible, criteria to decide on the classification of variants. It proposes a work system through a series of weighted criteria that allows a score to be made to decide on the greater or lesser probability of pathogenicity of the finding, with criteria in favor and criteria against:
Very strong pathogenicity criterionPVS1 null variant (nonsense, frame change, canonical ± 1 or 2 splice sites, start codon, single or multi-exon deletion) in a gene in which LOF is a known disease mechanism.
Strong pathogenicity criterionPS1 Same change of amino acids as in a previously established pathogenic variant, regardless of nucleotide change.
PS2 De novo (both maternity and paternity confirmed) in a patient with the disease and no family history.
PS3 Well-established in vitro or in vivo functional studies supporting a deleterious effect on the gene or gene product.
PS4 The prevalence of the variant in affected individuals increases significantly as compared to the prevalence in controls
PP1 (Strong Evidence) Co-segregation with disease in several affected family members in a gene definitely known to cause the disease
Moderate pathogenicity criteriaPM1 Located at a critical and/or mutational hotspot and well-established functional domain (eg, active site of an enzyme) with no benign variation.
PM2 Absent from controls (or extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium
PM3 For recessive disorders, detected in trans with a pathogenic variant
PM4 Protein length changes as a result of in-frame deletions/insertions in a non-repeat variant region
PM5 New sense change in an amino acid residue where a different sense change determined as a pathogen has been seen
PM6 Supposed de novo, but without confirmation of paternity and maternity
Mild pathogenicity criterionPP1 (Moderate evidence) Co-segregation with disease in multiple affected families members of a gene definitely known to cause the disease
PP1 Co-segregation with disease in multiple affected family members in a gene definitely known to cause the disease
PP2 Missense variant in a gene that has a low benign missense rate of variation and in which missense variants are a common disease mechanism
PP3 Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary impact, splicing, etc.)
PP4 The phenotype or family history of the patient is highly specific for a disease with a single genetic etiology.
PP5 A reliable source recently reports a variant as a pathogen, but the laboratory does not have evidence to conduct an independent evaluation
Moderate criterion of benignityBP1 variant missense in a gene for which is known that mainly truncating variants cause the disease
BP2 Observed in trans with a pathogenic variant for a fully penetrating dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern
BP3 In-frame deletions/insertions in a repeating region with no known function
BP4 Several lines of computational evidence suggest that there is no impact on the gene or gene product (conservation, evolution, splicing impact, etc.)
BP5 Variant found in a case with an alternative molecular basis for the disease
BP6 A reliable source recently reports a variant as benign, but the evidence is not available to the laboratory for independent evaluation.
BP7 A synonymous (silent) variant for which splice prediction algorithms do not predict any impact on the splice consensus sequence or creation of a new splice site and the nucleotide is not highly conserved
Strong criterion of benignityBS1 Allelic frequency is higher than expected for the disorder
BS2 Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with expected full penetrance at a young age
BS3 Well established in vitro or in vivo functional studies show no detrimental effects on protein splicing or function
BS4 Lack of segregation in affected members of a family
BA1 Allelic frequency is >5% in Exome Sequencing Project, 1000 Genome Project or Exome Aggregation Consortium
Our variant meets the criteria for strong evidence of PS4 pathogenicity (the prevalence of the variant is significantly higher in affected individuals compared to the prevalence in controls); of moderate evidence of pathogenicity PM1 (located in a well-established functional domain without benign variants) and PM2 (absent in controls or extremely low frequency in population databases); evidence supporting pathogenicity PP1 (co-segregation of disease in multiple affected family members in that gene), PP3 (various in silico computational predictors support a deleterious effect on the gene), and PP4 (phenotype or family history is highly specific for a disease with a unique genetic etiology); which allows it to be classified according to the algorithms of the guide as pathogenic or class 3.
The clinical impact of this missense variant is not different from all variants in the PKD1 gene considered globally; however, renal survival is superior when compared to those with a premature stop codon22 (stop codon 53.3 ± 0.56 vs. 58.5 ± 0.77 years; p < 0.05).
The diagnosis was made late, 29 years on average, and in more than half of the cases after having already an offspring. Although having a family history of the disease is one of the main reasons for making the diagnosis, in our study a large number of individuals, between 6–18/family, are still not investigated and some may be affected. This is because it has not been possible to establish a family connection between the 4 family trees, but they are located in the same geographical area, it makes us suspect that there was a founder effect (Table 3).
Criteria for considering a pathogenic variant. In bold the criteria that the variant described in our families meets.
| Criteria for Classifying Pathogenic Variants | Strong evidence of pathogenicity | PS4 | The prevalence of the variant in affected individuals is significantly increased compared to the prevalence in controls |
| Moderate evidence of pathogenicity | PM1 | Located in a mutational hot spot and/or critical and well-established functional domain (eg. Active site of an enzyme) without benign variation | |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes or ExAC | ||
| Supporting evidence of pathogenicity | PP1 | Co-segregation with disease in multiple affected family members in a gene definitively known to cause the disease | |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | ||
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | ||
| Criteria for Classifying Benign Variants | Supporting evidence of benign impact | BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease |
| Pathogenic | Class 1 | Very Strong (PVS1) AND | a. ≥1 Strong (PS1–PS4) OR b. ≥2 Moderate (PM1–PM6) OR c.1 Moderate (PM1–PM6) and 1 Supporting (PP1–PP5) OR d. ≥2 Supporting (PP1–PP5) |
| Class 2 | ≥2 Strong (PS1–PS4) | ||
| Class 3 | |||
| 1 Strong (PS1–PS4) AND | |||
| a. ≥3 Moderate (PM1–PM6) OR b.2 Moderate (PM1–PM6) AND ≥ 2 Supporting (PP1–PP5) OR c.1 Moderate (PM1–PM6) AND ≥ 4 Supporting (PP1–PP5) |
This table is excerpted from the 2015 American college guide.
Our work indicates 1) that the variant c.7292T> A in the PKD1 gene is pathogenic and therefore must be classified as such and 2) that its distribution in the Alpujarra region of Granada suggests a founder effect. In situations like this, segregation studies are necessary to characterize pathogenic variants not yet described and facilitate reproductive options such as TGP.
FinancingThis work has been funded by the José Luis Castaño-SEQC Foundation, 2018-2019 post-residency scholarship.
Conflict of interestsThe authors declare that they have no conflict of interests.
Please cite this article as: García-Rabaneda C, Martínez-Atienza M, Morales-García AI, Poyatos-Andújar A, García-Linares S, Bellido-Díaz ML et al. Nueva mutación asociada a poliquistosis renal autosómica dominante con efecto fundador localizada en la Alpujarra de Granada. Nefrologia. 2020;40:536–542.
Grupo de Estudio de la Enfermedad Poliquística Autosómica Dominante (GEEPAD).
Asociación Amigos del Riñón.
Instituto de Investigación Biosanitaria de Granada.


AL DÍA


- Home
- All contents
- Publish your article
- About the journal
- Metrics
- Open access