

Demostrar que la variante no descrita en el gen PKD1 c.7292T>A, identificada en cuatro familias de la comarca de la Alpujarra de Granada, es la causante de la poliquistosis renal autosómica dominante (PQRAD). Esta variante consiste en una sustitución transversión de timina (T) por adenina (A) que a nivel de la proteína policistina 1 produce un cambio de leucina (Leu/L) por glutamina (Gln/Q) en la posición 2431 (p.Leu2431Gln).
MétodoRegistramos variables sociodemográficas y clínicas a través de la realización de historias clínicas, árboles genealógicos, ecografías y estudios genéticos a individuos afectos y sanos pertenecientes a estas familias en el contexto del estudio de segregación.
ResultadosTodos los individuos afectados portaban en heterocigosis la variante c.7292T>A, mientras que los individuos sanos no la portaron. En las familias estudiadas, el 62,9% eran mujeres. El diagnóstico de PQRAD se realizó a los 29,3 ± 15,82 años de edad, después de haber tenido el primer hijo en el 64,8%. Los motivos principales de diagnóstico de la enfermedad fueron antecedentes familiares y episodios de hematuria. El inicio de tratamiento renal sustitutivo (TRS) se produjo a la edad de 55,8 ± 7,62 años (rango 44-67), y el éxitus a los 63 ± 92,2 años (rango 48-76), siendo la causa desconocida, cardiovascular e insuficiencia renal las más frecuentes; la mediana de supervivencia renal se estableció a los 58,5 ± 0,77 años y la mediana de supervivencia del paciente a los 67 ± 3,54 años. No observamos diferencias en la supervivencia del riñón y del paciente según el sexo. De los pacientes fallecidos, el 52,2% necesitaron TRS y el 94,4% tenían algún grado de insuficiencia renal (IR).
ConclusionesLa variante c.7292T>A en el gen PKD1 es responsable de la enfermedad y su distribución en la comarca de la Alpujarra de Granada sugiere un efecto fundador. En la PQRAD es necesario realizar estudios de segregación que ayuden a reclasificar variantes genéticas, en este caso de indeterminada a patogénica.
To demonstrate that the variant not described in PKD1 gene c.7292T> A, identified in four families from the Alpujarra in Granada, is the cause of autosomal dominant polycystic kidney disease (ADPKD). This variant consists of a transversion of thymine (T) by adenine (A) that at the level of the Polycystin 1 protein produces a change of leucine (Leu / L) by Glutamine (Gln / Q) in position 2431 (p.Leu2431Gln).
MethodSociodemographic and clinical variables were registered using clinical histories, genealogical trees, ultrasounds and genetic analysis to ADPKD and healthy individuals belonging to these families in the context of segregation study.
ResultsAll PKD individuals carried the c.7292T>A variant in heterozygosis, whereas healthy ones did not. Among all ADPKD patients, 62.9% were women. ADPKD diagnosis was made at 29.3 ± 15.82 years, after having the first child in 64.8%. The main reasons for diagnosis were family history and hematuria episodes. The onset of renal replacement therapy (RRT) occurred at 55.8 ± 7.62 years (range 44-67), and death at 63 ± 92.2 years (range 48-76), being the cause unknown, cardiovascular and insufficiency kidney the most frequent; the median of renal survival was established at 58.5 ± 0.77 years and the median survival of patients at 67.2 ± 3.54 years. No differences in kidney and patient survivals were observed according to sex. Among deceased patients, 52.2% required RRT and 94.4% suffered from renal failure.
ConclusionsThe variant c.7292T>A in PKD1 gene is responsible for the disease, and its distribution in the Alpujarra region of Granada suggests a founder effect. In ADPKD it is necessary to perform segregation studies that help us to reclassify genetic variants, in this case from indeterminate to pathogenic.
La poliquistosis renal autosómica dominante (PQRAD), clasificada con los códigos internacionales 753.12, 753.13 (CIE-9) y Q61.2, Q61.3 (CIE-10), es la nefropatía hereditaria más común que ocasiona fallo renal y necesidad de tratamiento renal sustitutivo (TRS). Cursa con aparición y crecimiento progresivo de quistes renales que sustituyen la totalidad del parénquima renal normal acompañado de fibrosis e inflamación intersticial1. Es una enfermedad monogénica y/o bigénica2 que puede afectar al riñón y a otros órganos y originar hipertensión arterial, infecciones urinarias, dolor lumbar o abdominal, hematuria, nefrolitiasis y fallo renal, así como quistes hepáticos, en páncreas y epidídimo, plexo coroidal y aracnoideos, aneurismas cerebrales, anomalías cardíacas y hernias abdominales, entre otras3. La intensidad de las manifestaciones es variable incluso entre individuos de una misma familia4.
La prevalencia no está bien determinada, ya que no disponemos de registros oficiales específicos, aunque estudios recientes la señalan entre 4,76-9,14 casos/10.000 nacidos5. Nuestro registro, compuesto por 298 familias y 1.164 pacientes identificados en el ámbito sanitario de Granada, identifica entre 16-31 nuevos casos/año (periodo 2000-2018), con una incidencia media de 2,34 casos/105 personas/año6, datos que concuerdan con lo informado en el estudio Else-Kroener-Fresenius-ADPKD en el suroeste de Alemania7.
La mayoría de los casos se deben a variantes en el gen PKD1 (16p13.3), y en menor proporción en los genes PKD2 (4q22.1), GANAB (11q12.3)8 y DNAJB11 (3q27.3), existiendo pocos casos de afectación combinada en los genes PKD1 y PKD29. Estos genes codifican las proteínas policistina 1 (PKD1), policistina 2 (PKD2) y a la subunidad α de la glucosidasa II (GANAB), muy ubicuas en el organismo, aunque forman parte principalmente del cilio primario10. Su alteración va a provocar una disminución del calcio e incremento de AMPc intracelulares, alteraciones de la proliferación celular y de la polaridad, aumento de la secreción de fluidos e incremento de la matriz extracelular, todos ellos implicados en la quistogénesis, aunque el mecanismo exacto aún se desconoce10,11.
Nuestro trabajo pretende demostrar que la nueva variante c.7292T>A identificada en el gen PKD1 es responsable de la PQRAD en 4 familias de la Alpujarra de Granada, aparentemente no relacionadas mediante árbol genealógico, así como identificar la existencia de un posible efecto fundador.
Material y métodosSe han realizado historias clínicas, árboles genealógicos y ecografías a miembros pertenecientes a 4 familias afectas por PQRAD oriundas de la comarca geográfica de la Alpujarra de Granada, donde hay censadas 25.000 personas: una familia es oriunda de Órgiva, con 5.570 habitantes y las otras tres de Pórtugos, con 403 habitantes12.
Se realizaron estudios genéticos mediante secuenciación masiva (NGS) a personas con diagnóstico de PQRAD13. Se identificó en heterocigosis la variante c.7292T>A (p.Leu2431Gln) en el gen PKD1, no descrita en las bases de datos ni en la literatura. Por otra parte, el análisis in silico resultó contradictorio: los programas PoliPhen2, SIFT y Mutation Taster señalan un efecto deletéreo mientras que Align GVGD señaló un efecto benigno. Esta situación obligó a planificar el estudio de segregación: realizamos análisis genéticos en familiares afectos (secuenciación completa del gen PKD1 o estudio de mutación puntual conocida, según el caso) y en familiares sanos (estudio de mutación puntual conocida).
En cada familia hemos estudiado individuos afectos y sanos. Todos los participantes fueron informados y firmaron el consentimiento informado; los realizados en menores de edad fueron autorizados por sus progenitores.
Los estudios genéticos se realizaron en el Instituto de Medicina Genómica (IMEGEN) empleando el panel de secuenciación masiva (NGS) NextGeneDx®, que incluye los genes PKD1, PKD2 y GANAB, siguiendo la siguiente metodología:
- 1.
Extracción de ADN genómico a partir de los linfocitos de la muestra sanguínea.
- 2.
Preparación de librerías mediante el kit Nextera XT (Ilumina).
- 3.
Secuenciación de las librerías (2 × 150) en el secuenciador MiSeq (Ilumina).
- 4.
Análisis bioinformático de las secuencias obtenidas.
- 5.
Confirmación de variantes por secuenciación Sanger.
Para evaluar la patogenicidad de las variantes encontradas se utilizaron bases de datos poblacionales como la Human Gene Mutation Database (HGMD), Clinvar, GenomeAD, LOVD y, la que ofrece mayor información específica, la ADPKD Mutation Database, que identifica 2.601 cambios en la estructura del gen PKD1, entre silenciosos, patogénicos y de significado clínico incierto o indeterminado14.
Para las mutaciones puntuales en pacientes con mutación ya conocida, se realizó secuenciación Sanger por el laboratorio Imegen.
Registramos variables sociodemográficas, clínicas y otras relativas al impacto de la mutación sobre riñón y paciente mediante estudios de supervivencia. Empleamos los programas Geno-Pro para realizar los árboles genealógicos y el paquete estadístico SPSS 15.0. Los datos se expresan en términos de media ± DT, rango, mediana y porcentaje, según el caso. Los estudios de supervivencia renal y del paciente se realizaron con la prueba de Kaplan-Meier, considerándolo significativo cuando p < 0,05.
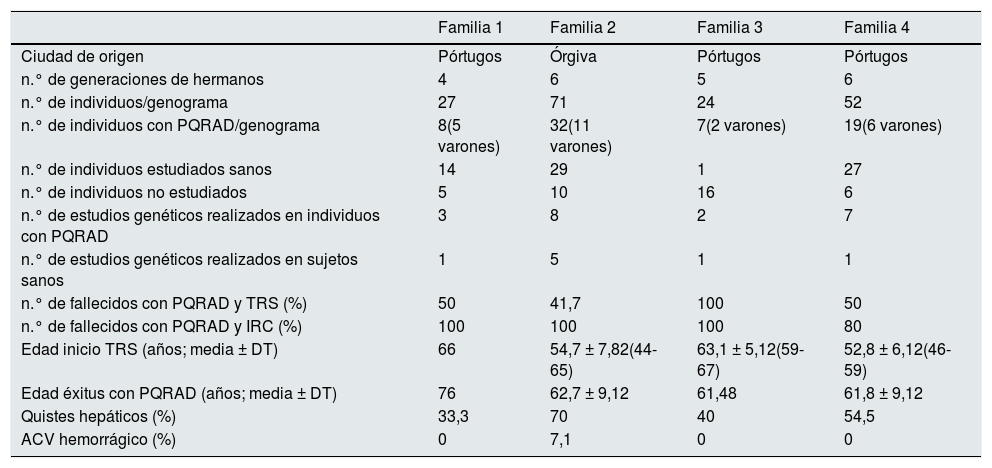
ResultadosEn la tabla 1 se resumen características sociodemográficas, clínicas y genéticas de la población estudiada. Por motivos de accesibilidad a la consulta, realizamos un estudio genético en 20 de los 66 pacientes diagnosticados con PQRAD mediante criterios ecográficos de de Ravine y Pei modificados13 en estas familias (30,3%): 8 mediante secuenciación masiva y 12 mediante secuenciación Sanger para el estudio de mutaciones puntuales conocidas. El análisis genético reveló la presencia de la variante c.7292T>A (p.Leu2431Gln) en todos los afectos. Asimismo, se estudiaron 8 familiares sin evidencia clínica y ecográfica de la enfermedad y en ninguno se identificó la variante.
Características sociodemográficas, clínicas y genéticas de las familias (IRC: insuficiencia renal crónica (eFG < 60 mL/min); TRS: tratamiento renal sustitutivo)
| Familia 1 | Familia 2 | Familia 3 | Familia 4 | |
|---|---|---|---|---|
| Ciudad de origen | Pórtugos | Órgiva | Pórtugos | Pórtugos |
| n.° de generaciones de hermanos | 4 | 6 | 5 | 6 |
| n.° de individuos/genograma | 27 | 71 | 24 | 52 |
| n.° de individuos con PQRAD/genograma | 8(5 varones) | 32(11 varones) | 7(2 varones) | 19(6 varones) |
| n.° de individuos estudiados sanos | 14 | 29 | 1 | 27 |
| n.° de individuos no estudiados | 5 | 10 | 16 | 6 |
| n.° de estudios genéticos realizados en individuos con PQRAD | 3 | 8 | 2 | 7 |
| n.° de estudios genéticos realizados en sujetos sanos | 1 | 5 | 1 | 1 |
| n.° de fallecidos con PQRAD y TRS (%) | 50 | 41,7 | 100 | 50 |
| n.° de fallecidos con PQRAD y IRC (%) | 100 | 100 | 100 | 80 |
| Edad inicio TRS (años; media ± DT) | 66 | 54,7 ± 7,82(44-65) | 63,1 ± 5,12(59-67) | 52,8 ± 6,12(46-59) |
| Edad éxitus con PQRAD (años; media ± DT) | 76 | 62,7 ± 9,12 | 61,48 | 61,8 ± 9,12 |
| Quistes hepáticos (%) | 33,3 | 70 | 40 | 54,5 |
| ACV hemorrágico (%) | 0 | 7,1 | 0 | 0 |
En estas 4 familias, el 62,9% de los afectados eran mujeres. El diagnóstico se realizó a los 29,3 ± 15,82 años de edad (rango edad 1-59), en el 64,8% después de haber tenido un primer hijo. La presencia de antecedentes familiares y la hematuria fueron las dos causas principales que condujeron al diagnóstico. En los familiares afectos, identificamos quistes hepáticos en el 59,3%, historia de accidente cerebrovascular hemorrágico en el 3,3% y quistes en epidídimo u ovario en el 13%. La edad de inicio del tratamiento renal sustitutivo (TRS) fue a los 55,8 ± 7,62 años (rango edad 44-67); el éxitus se produjo a los 63,1 ± 9,32 años (rango edad 48-76), siendo la causa desconocida, cardiovascular e insuficiencia renal las más frecuentes. La mediana de supervivencia renal fue de 58,5 ± 0,77 años y la mediana de supervivencia del paciente fue de 67,2 ± 3,54 años, sin encontrar diferencias significativas en función del sexo. De los pacientes fallecidos, el 52,2% necesitaron TRS y el 94,4% tenían algún grado de insuficiencia renal (IR).
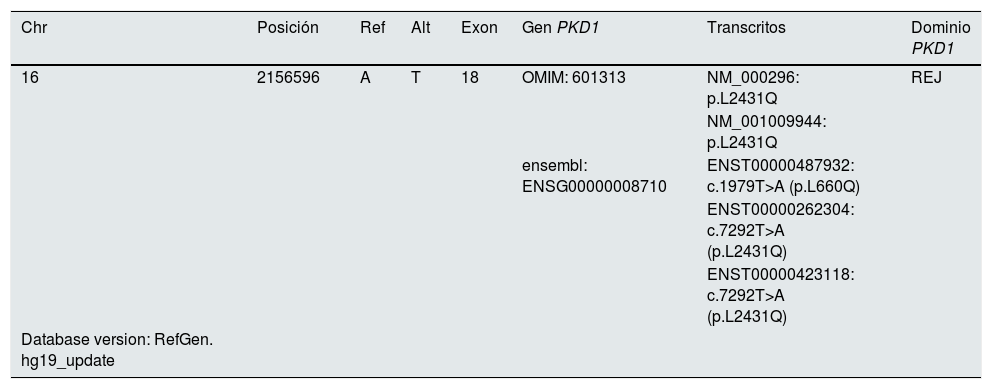
DiscusiónNuestra variante, no identificada en los registros de variantes génicas humanas generales ni específicas, se encuentra localizada en el exón 18 en la posición cromosómica chr16:2156596 donde se produce una sustitución transversión de adenina a timina que predice una mutación tipo cambio de sentido (missense) con la modificación de leucina (L) por glutamina (Q) en el codón 2431 (c.7292T>A, L2431Q) (tabla 2).
Caracterización de la variante según diferentes tránscritos del gen PKD1
| Chr | Posición | Ref | Alt | Exon | Gen PKD1 | Transcritos | Dominio PKD1 |
|---|---|---|---|---|---|---|---|
| 16 | 2156596 | A | T | 18 | OMIM: 601313 | NM_000296: p.L2431Q | REJ |
| NM_001009944: p.L2431Q | |||||||
| ensembl: ENSG00000008710 | ENST00000487932: c.1979T>A (p.L660Q) | ||||||
| ENST00000262304: c.7292T>A (p.L2431Q) | |||||||
| ENST00000423118: c.7292T>A (p.L2431Q) | |||||||
| Database version: RefGen. hg19_update |
Este cambio se ubica en la región extramembranosa, dominio REJ de la policistina 1 (PKD1). El dominio en PKD1 tiene una longitud de casi 1.000 aminoácidos comprendidos entre los exones 15 y 27. Probablemente esté compuesto por múltiples dominios estructurales. Hay seis residuos de cisteína completamente conservados que pueden formar puentes disulfuro15. La función de este dominio no está bien establecida, aunque se ha relacionado con la regulación del flujo de iones calcio16. Algunos autores sugieren que se trata de una región esencial para la actividad biológica de la policistina-117. El dominio REJ se encuentra en la proximidad del dominio GPS (sitio proteolítico acoplado a la proteína G), que es esencial para la estructura y función del riñón. Algunos trabajos muestran que el dominio GPS requiere para su función de la integridad de la región REJ. En la base de datos PKD de la Clínica Mayo hay descritas alrededor de 65 variantes missense clasificadas como causales de enfermedad en esta región15. En esta misma región y muy próximas en la secuencia de la proteína están descritas otras variantes missense clasificadas como probablemente patogénicas tales como c.7300C>T, p.Arg2434Trp (R2434W)18, c.7301G>A, p.Arg2434Gln (R2434Q)19 y p.Leu2433Arg (L2433R)20.
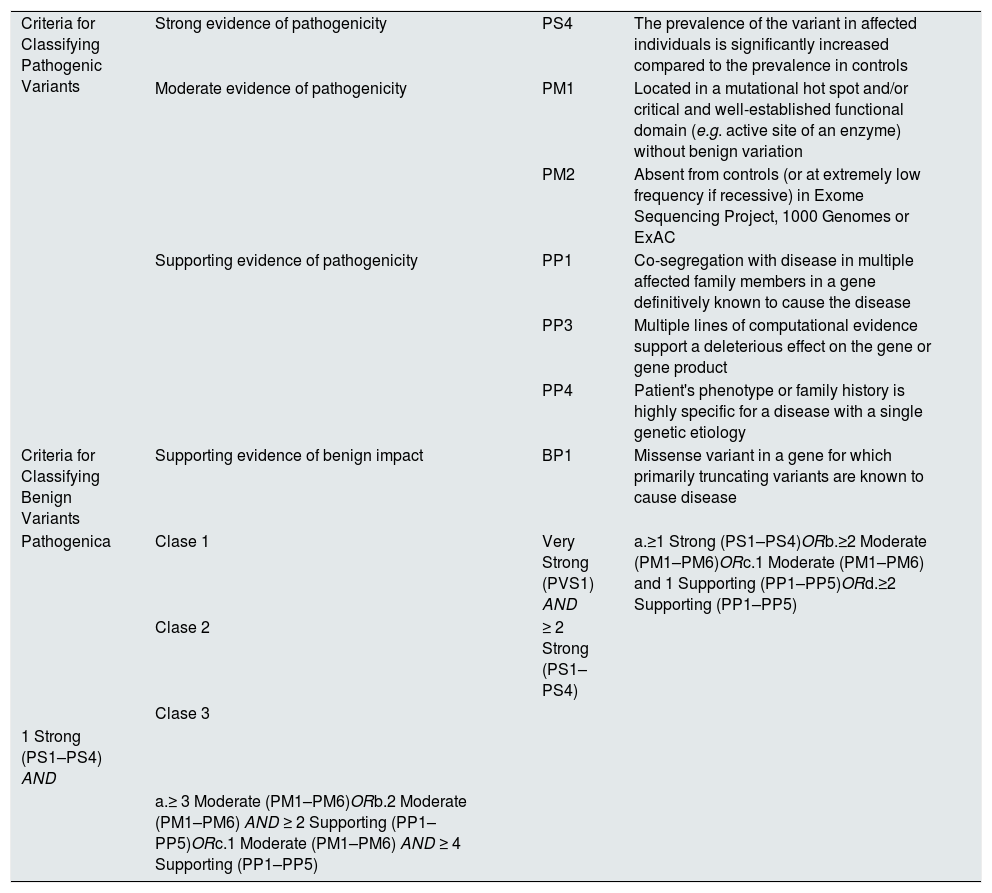
Unos predictores in silico la clasifican como deletérea y otro como benigna. En las bases de datos ExAC, ESP5400 y 1000 Genomes no está registrada, lo que indica su baja frecuencia poblacional. Esta contradicción de predictores hace que la variante se clasifique como indeterminada. El estudio de segregación es contundente: la variante la portan 20 personas afectadas y ninguna de los 8 individuos sanos estudiados. Es decir, la variante cosegrega con la enfermedad. Para clasificar una variante se recomienda seguir los criterios de la American College of Medical Genetics and Genomics21. En 2015, se redactó esta guía de consenso para intentar objetivar, en la medida de lo posible, criterios para decidir sobre la clasificación de variantes. Propone una sistemática de trabajo mediante una serie de criterios ponderados que permite realizar un score (puntaje) para decidir sobre la mayor o menor probabilidad de patogenicidad del hallazgo, existiendo criterios a favor y criterios en contra:
Criterio muy fuerte de patogenicidadVariante nula de PVS1 (sin sentido, cambio de marco, canónico ± 1 o 2 sitios de empalme, codón de iniciación, eliminación simple o multiexón) en un gen en el que la LOF es un mecanismo de enfermedad conocido
Criterio fuerte de patogenicidadPS1 Mismo cambio de aminoácidos que una variante patógena establecida previamente, independientemente del cambio de nucleótido
PS2 De novo (tanto la maternidad como la paternidad confirmada) en un paciente con la enfermedad y sin antecedentes familiares
PS3 Estudios funcionales in vitro o in vivo bien establecidos que respalden un efecto dañino sobre el gen o producto genético
PS4 La prevalencia la variante en los individuos afectados aumenta significativamente en comparación con la prevalencia en los controles
PP1 (Pruebas sólidas) Cosegregación con enfermedad en varios miembros de la familia afectados en un gen definitivamente conocido como causante de la enfermedad
Criterio moderado de patogenicidadPM1 Ubicado en un punto crítico mutacional y/o crítico y un dominio funcional bien establecido (p. ej., sitio activo de una enzima) sin variación benigna
PM2 Ausente de los controles (o con una frecuencia extremadamente baja si es recesivo) en Exome Sequencing Project, 1000 Genomes Project o Exome Aggregation Consortium
PM3 Para trastornos recesivos, detectados en trans con una variante patógena
PM4 La longitud de la proteína cambia como resultado de deleciones/inserciones en el marco en una región sin repetición variantes
PM5 Nuevo cambio de sentido en un residuo de aminoácido donde se ha visto un cambio de sentido diferente determinado como patógeno
PM6 Supuesto de novo, pero sin confirmación de paternidad y maternidad
Criterio leve de patogenicidadPP1 (Evidencia moderada) Cosegregación con enfermedad en múltiples familias afectadas miembros de un gen definitivamente conocido como causante de la enfermedad
PP1 Cosegregación con enfermedad en múltiples miembros de la familia afectados en un gen definitivamente conocido como causante de la enfermedad
PP2 Missense variante en un gen que tiene una baja tasa de variación benigna de missense y en la que variantes de missense son un mecanismo común de la enfermedad
PP3 Múltiples líneas de evidencia computacional apoyan un efecto perjudicial sobre el gen o producto genético (conservación, impacto evolutivo, empalme, etcétera)
PP4 El fenotipo o la historia familiar del paciente es altamente específico para una enfermedad con una única etiología genética.
PP5 Una fuente confiable recientemente reporta una variante como patógena, pero el laboratorio no cuenta con pruebas para realizar una evaluación independiente
Criterio moderado de benignidadBP1 Variante de missense en un gen para el que se sabe que las variantes principalmente truncantes causan enfermedad
BP2 Observada en trans con una variante patógena para un gen/trastorno dominante completamente penetrante u observada en cis con una variante patógena en cualquier patrón de herencia
BP3 Eliminaciones/inserciones en marco en una región repetitiva sin una función conocida
BP4 Varias líneas de evidencia computacional sugieren que no hay impacto en el gen o producto genético (conservación, evolución, impacto de empalme, etcétera)
BP5 Variante encontrada en un caso con una base molecular alternativa para la enfermedad
BP6 Una fuente confiable recientemente reporta una variante como benigna, pero la evidencia no está disponible para el laboratorio para que este realice una evaluación independiente
BP7 Una variante sinónima (silenciosa) para la cual los algoritmos de predicción de empalme no predicen ningún impacto en la secuencia de consenso de empalme ni en la creación de un nuevo sitio de empalme y el nucleótido no está muy conservado
Criterio fuerte de benignidadBS1 La frecuencia alélica es mayor de lo esperado para el trastorno
BS2 Observado en un individuo adulto sano para un trastorno recesivo (homocigoto), dominante (heterocigoto) o ligado al X (hemicigótico), con penetrancia completa esperada a una edad temprana
BS3 Los estudios funcionales in vitro o in vivo bien establecidos muestran que no hay efectos perjudiciales sobre la función o el empalme de la proteína
BS4 Falta de segregación en los miembros afectados de una familia
BA1 La frecuencia alélica es > 5% en el Exome Sequencing Project, 1000 Genome Project o Exome Aggregation Consortium
Nuestra variante cumple con criterios de fuerte evidencia de patogenicidad PS4 (la prevalencia de la variante es significativamente mayor en individuos afectos comparada con la prevalencia en controles); de moderada evidencia de patogenicidad PM1(ubicado en un dominio funcional bien establecido sin variantes benignas) y PM2 (ausente en los controles o con frecuencia extremadamente baja en las bases de datos poblacionales); pruebas que apoyan la patogenicidad PP1 (cosegregación de la enfermedad en múltiples miembros de la familia afectados en ese gen), PP3 (varios predictores computacionales in silico apoyan un efecto deletéreo sobre el gen) y PP4 (el fenotipo o el historial familiar es muy específico para una enfermedad con una etiología genética única); lo que permite clasificarla según los algoritmos de la guía como patogénica o clase 3.
El impacto clínico de esta variante missense no es diferente respecto a todas las variantes en el gen PKD1 consideradas de manera global; sin embargo, la supervivencia renal es superior si la comparamos con aquellos que portan un codón de parada prematuro22 (codón de parada 53,3 ± 0,56 vs. 58,5 ± 0,77 años; p < 0,05).
El diagnóstico se realizó de manera tardía, 29 años de media, y en más de la mitad de las ocasiones tras haber tenido ya descendencia. Por otra parte, si bien tener antecedentes familiares de la enfermedad es uno de los principales motivos para realizar el diagnóstico, en nuestro estudio un nutrido número de individuos, entre 6-18/familia, se encuentran aún sin investigar y algunos pueden estar afectados. Debido a que no se ha podido establecer una conexión familiar entre los 4 árboles genealógicos, pero se ubican en la misma área geográfica, hace sospechar que existió un efecto fundador (tabla 3).
Criterios para considerar una variante patogénica. En negrita los criterios que cumple la variante descrita en nuestras familias
| Criteria for Classifying Pathogenic Variants | Strong evidence of pathogenicity | PS4 | The prevalence of the variant in affected individuals is significantly increased compared to the prevalence in controls |
| Moderate evidence of pathogenicity | PM1 | Located in a mutational hot spot and/or critical and well-established functional domain (e.g. active site of an enzyme) without benign variation | |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes or ExAC | ||
| Supporting evidence of pathogenicity | PP1 | Co-segregation with disease in multiple affected family members in a gene definitively known to cause the disease | |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | ||
| PP4 | Patient's phenotype or family history is highly specific for a disease with a single genetic etiology | ||
| Criteria for Classifying Benign Variants | Supporting evidence of benign impact | BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease |
| Pathogenica | Clase 1 | Very Strong (PVS1) AND | a.≥1 Strong (PS1–PS4)ORb.≥2 Moderate (PM1–PM6)ORc.1 Moderate (PM1–PM6) and 1 Supporting (PP1–PP5)ORd.≥2 Supporting (PP1–PP5) |
| Clase 2 | ≥ 2 Strong (PS1–PS4) | ||
| Clase 3 | |||
| 1 Strong (PS1–PS4) AND | |||
| a.≥ 3 Moderate (PM1–PM6)ORb.2 Moderate (PM1–PM6) AND ≥ 2 Supporting (PP1–PP5)ORc.1 Moderate (PM1–PM6) AND ≥ 4 Supporting (PP1–PP5) |
Esta tabla está extraída de la guía de la American college de 2015.
Nuestro trabajo señala 1) que la variante c.7292T>A en el gen PKD1 es patogénica y por ende debe ser clasificada como tal y 2) que su distribución en la comarca de la Alpujarra de Granada sugiere un efecto fundador. En situaciones como esta, los estudios de segregación son necesarios para caracterizar variantes patogénicas aún no descritas y facilitar opciones reproductivas como el TGP.
FinanciaciónEl presente trabajo ha sido financiado por la fundación José Luis Castaño-SEQC, beca post-residencia 2018-2019.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Grupo de Estudio de la Enfermedad Poliquística Autosómica Dominante (GEEPAD).
Asociación Amigos del Riñón.
Instituto de Investigación Biosanitaria de Granada.


AL DÍA

