











In recent years there has been a reclassification of hereditary tubulointerstitial renal diseases. The old concepts of nephronoptisis or medullary cystic disease have been reordered based on the discovery of new genes. The 2015 KDIGO guidelines proposed a unification of terminology, diagnostic criteria and monitoring. So far 4 genes causing autosomal dominant tubulointerstitial kidney disease have been described: MUC1, UMOD, HNF1B and REN. Although the mutation in each of them causes distinctive features in how they present, all have in common the progressive tubulointerstitial damage and renal fibrosis. In this article, we present a review of the guidelines and the literature, and some practical recommendations for dealing with this disease.
En los últimos años ha habido una reclasificación de las nefropatías tubulointersticiales de base genética. Los antiguos conceptos de nefronoptisis o enfermedad quística medular han sido reordenados con base en el hallazgo de nuevos genes. Las guías KDIGO del 2015 proponen una unificación de terminología, unos criterios diagnósticos y de seguimiento. Hasta el momento se han descrito 4 genes causantes de la nefropatía tubulointersticial autosómica dominante: MUC1, UMOD, HNF1B y REN. Aunque la mutación en cada uno de los genes produce unos rasgos diferenciales en la forma de presentación, todas las formas tienen en común el progresivo daño túbulo-intersticial y la fibrosis renal. En este artículo, se pretende una revisión de las guías, de la literatura y ofrecer unas recomendaciones prácticas para el manejo de esta enfermedad.
The term interstitial nephropathies includes diseases that predominantly affect the renal interstitium; although, to various degree all elements of the renal parenquima (glomeruli, tubules and vessels) may also be involved. Since renal tubular cells are often damaged, some authors rather use the term tubulointerstitial nephropathies.1 The progression of renal disease is clearly related to the tubulointertitial damage.2
Within tubulointerstitial nephropathies, the are familiar forms with a very heterogeneous clinical profile even within the same family.3 Corticomedullary cysts are present in several families and were clearly differentiated by the age of onset, then the concept of “nephronoptisis complex – cystic medullary disease” was coined.4,5 Nephronoptisis defines childhood forms with autosomal recessive inheritance, and the genes initially described were NPHP1 (nephrocystin protein) and INVS (inversin protein)6,7; up to 19 genes causing different forms of nephronoptisis have been described to date.7 The term medullary cystic disease was applied to adult forms with autosomal dominant inheritance, and the first gene identified was UMOD (uromodulin).8,9
More knowledge about the genes involved and the very inconstant presence of cysts requires changes in these concepts. Hopefully the clinical profile of this entity, with anodyne presentation and variable evolution, will be better defined.10
TerminologyThe term autosomal dominant tubulointerstitial kidney disease (KD) has recently been established by the KDIGO guidelines using the acronym ADTKD.11 Prior to this consensus, the nomenclature was non uniform being a source of confusion (Table 1).
Unification of new and old terminology.
| Gen mutation | New terminology | Old terminology |
|---|---|---|
| UMOD | ADTKD-UMOD (English) | UKD (uromodulin kidney disease) |
| NTAD-UMOD (Spanish) | UAKD (uromodulin-associated kidney disease)25,41 | |
| FJHN (familial juvenile hyperuricemic nephropathy)21 | ||
| MCKD2 (medullar cystic kidney disease type 2)8 | ||
| MUC1 | ADTKD-MUC1 (English) | MKD (mucin-1 kidney disease) |
| NTAD-MUC1 (Spanish) | MCKD1 (medullar cystic kidney disease type 1)13 | |
| REN | ADTKD-REN (English) | FJHN2 (familial juvenile hyperuricemic nephropathy type 2)17 |
| NTAD-REN (Spanish) | ||
| HNF1B | ADTKD-HNF1B (English) | MODY5 (maturity-onset diabetes mellitus of the young type 5)33 |
| NTAD-HNF1B (Spanish) | RCAD (renal cysts and diabetes syndrome) |
ADTKD: autosomal dominant tubulointerstitial kidney disease; NTAD: nefropatía tubulointersticial autosómica dominante (in Spanish).
The working group of the KDIGO guidelines decided to unify the terminologies and the clinical features of these rare inherited renal diseases,11 which have in common the tubulointerstitial fibrosis and slow progression to chronic end-stage renal disease (ESRD).1,12 The advantages of the new terminology are:
- •
Reveals the genetic origin of these diseases with an autosomal dominant inheritance pattern. It separates these diseases of the rest of the acquired chronic tubulointerstitial nephropathies, and nephronoptisis (of autosomal recessive inheritance).
- •
Summarizes the clinical characteristics of the disease caused by mutations in four different genes.
- •
Allows for clinical suspicion before histological or genetic diagnosis.
- •
Allows to differentiate these diseases from other dominant autosomal diseases of tubular origin (such as autosomal dominant polycystic kidney disease or distal tubular acidosis).
- •
It avoids some of the previous terminologies that could cause confusion, especially those that include the terms “cystic diseases” or “medullary cysts”.
- •
The terminology is simple and easy to use.
The penetrance of the disease is very high, close to 100% if patient lives long enough; however, severity and age of onset vary considerably among families and also within a family.11,13
The disease progresses slowly. The age of onset of ESRD is highly variable, from 25 to 70 years in patients with a mutation in the UMOD gene.3,14,15 The rate of decline in glomerular filtration is variable, and it depends on the mutated gene, but in general the decline in renal function is slow.3,13,16
Signs and symptoms are not specific:
- •
Proteinuria is negative or present in small amount (<1g/day).3,13,16
- •
Urinary sediment is usually normal or, exceptionally, with microhematuria.3,13,16
- •
The size of the kidneys is normal and decreases as the disease progresses. Renal cysts, which are usually located corticomedullary, are relatively frequent but the finding is not always constant and is often found in advanced stages of the disease.3,13,16
- •
In patients with a mutation in the UMOD gene, hyperuricemia may be found in at very variable age (ranging from 3 to 51 years).3,14
- •
Hypertension is common, but it does not appear early and usually is not severe.11
- •
Anemia occurs early and is out of proportion relative to the degree of renal failure; it is more severe when the disease is caused by mutation in the REN gene.17
This is a disease with tubulointerstitial damage however tubular functional abnormalities are not characteristic. There are urine concentration defects and abnormal proximal tubular reabsorption of uric acid in patients with UMOD mutations fundamentally. In experimental studies other damaged cationic transporters have been identified, but there is no clear evidence on patients clinical impact.18–20
HistologyThe clinical expression is poor and it progresses slowly so the diagnosis by renal biopsy is infrequent. They have interstitial fibrosis, tubular atrophy and normal glomeruli (Fig. 1). Progressive loss of the glomerular basement membrane is frequent and there is tubular dilatation with tubular microcystis.3,13,16,21,22 Immunofluorescence is negative. Electron microscopy does not usually contribute to the differential diagnosis. It can be observed an accumulation of the mutant uromodulin in the endoplasmic reticulum of the cells of the thick ascending limb of Henle's loop.23
Genetic defects and clinical presentation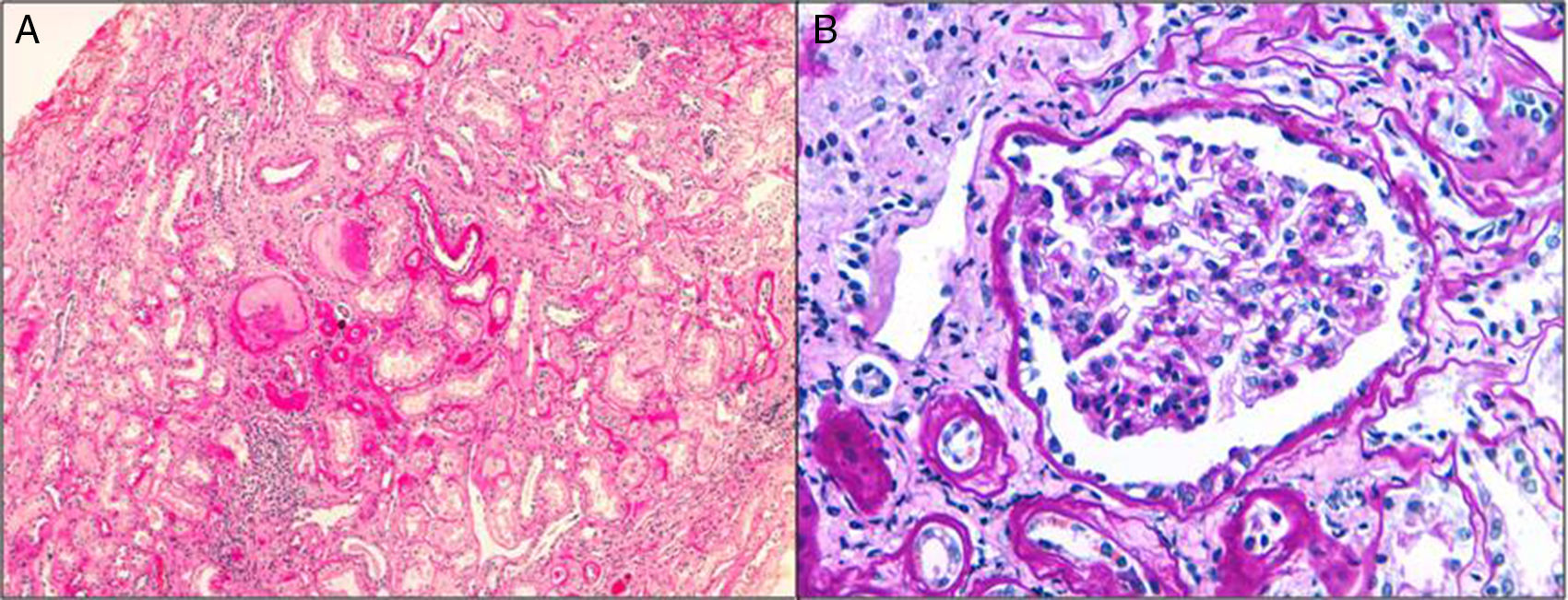
To date, there are 4 genes described that can be the cause of the disease (Table 2). Although the percentage of ADTKD caused by mutations in each genes is not clearly defined, it appears that mutations in UMOD and MUC1 would result in a higher percentage of cases of ADTKD than those caused by mutations in the REN or HNF1b genes.11
Characteristics of the different ADTKD.
| Name | Protein | Characteristics |
|---|---|---|
| ADTKD-UMOD | Codifies the protein uromodulin or Tamm-Horsfall19,42 | Inappropriate reduction of fractional excretion of Uric Acid uric (<5%) |
| Early hyperuricemia with or without gout episodes before CKD (non-pathognomonic)3 | ||
| Defects in urine concentration24 | ||
| Reduced urinary excretion of uromodulin24 | ||
| Occasionally, multiple cortical-medullary cysts3 | ||
| ADTKD-MUC1 | Codifies the protein mucin-130 | There are no specific characteristics other than tubulointerstitial fibrosis12,13 |
| Renal cysts may be present12,13 | ||
| ADTKD-HNF1B | Codifies the hepatic nuclear factor 1 beta16 | • Kidney: cortical and bilateral renal cysts, renal hypoplasia glomerulocystic disease, renal agenesia, renal hyperechogenicity in fetal and neonatal period, etc. |
| • Hypomagnesemia, hyperuricemia, impaired liver function tests 16 | ||
| • Extrarenal: MODY 5 type diabetes, genital malformations, atrophy of páncreas16 | ||
| ADTKD-REN | Codifies the protein renin17 | Anemia that is disproportionate for the degree of CKD. Early onset, in childhood/adolescence17,40 |
| Tendency to hyperkalemia and hyperuricemia17,40 | ||
| Low or normal blood pressure40 |
CKD: chronic kidney disease; MODY 5: maturity onset diabetes of the young; ADTKD: autosomal dominant tubulointerstitial kidney disease
The UMOD gene (chromosomal region 16p2) encodes for the protein uromodulin or Tamm-Horsfall, the most abundant protein in the urine, however the function of this protein has not been fully elucidated (it has been related to the impermeabilization of the distal tubule and a proinflammatory activity) produced in the epithelial cells of the ascending portion of the loop of Henle.19,24,25 Mutant uromodulin accumulates in the endoplasmic reticulum of cells so release and urinary excretion is decreased.25 Also, the intracellular traffic of the Na/K/2Cl (NKCC) cotransporter to the apical surface of the thick ascending limb of Henle is inhibited; consequently the sodium reabsorption is reduced and volume depletion may occur which favors the proximal reabsorption of uric acid.26
Clínical featuresHyperuricemia is frequent and, sometimes, with gouty crisis; hyperuricemia is due to the decrease in the fractional excretion of uric acid27 and it is disproportionate to the degree of renal failure; it may be present before the development of renal failure and may be observed in childhood and adolescence.3 End-stage renal disease occurs between 25 and 70 years and earlier in those with gout.21 Studies have shown that treatment of hyperuricemia with febuxostat slows down the progression of renal disease stage 3–4.28 Whether allopurinol slows the progression of renal disease is unclear.11,29Fig. 2 shows an example of a family with ADTKD due to UMOD gene mutation, where great intrafamily variability is observed.
Autosomal dominant tubulointerstitial nephropathy caused by mutations in the MUC1 gene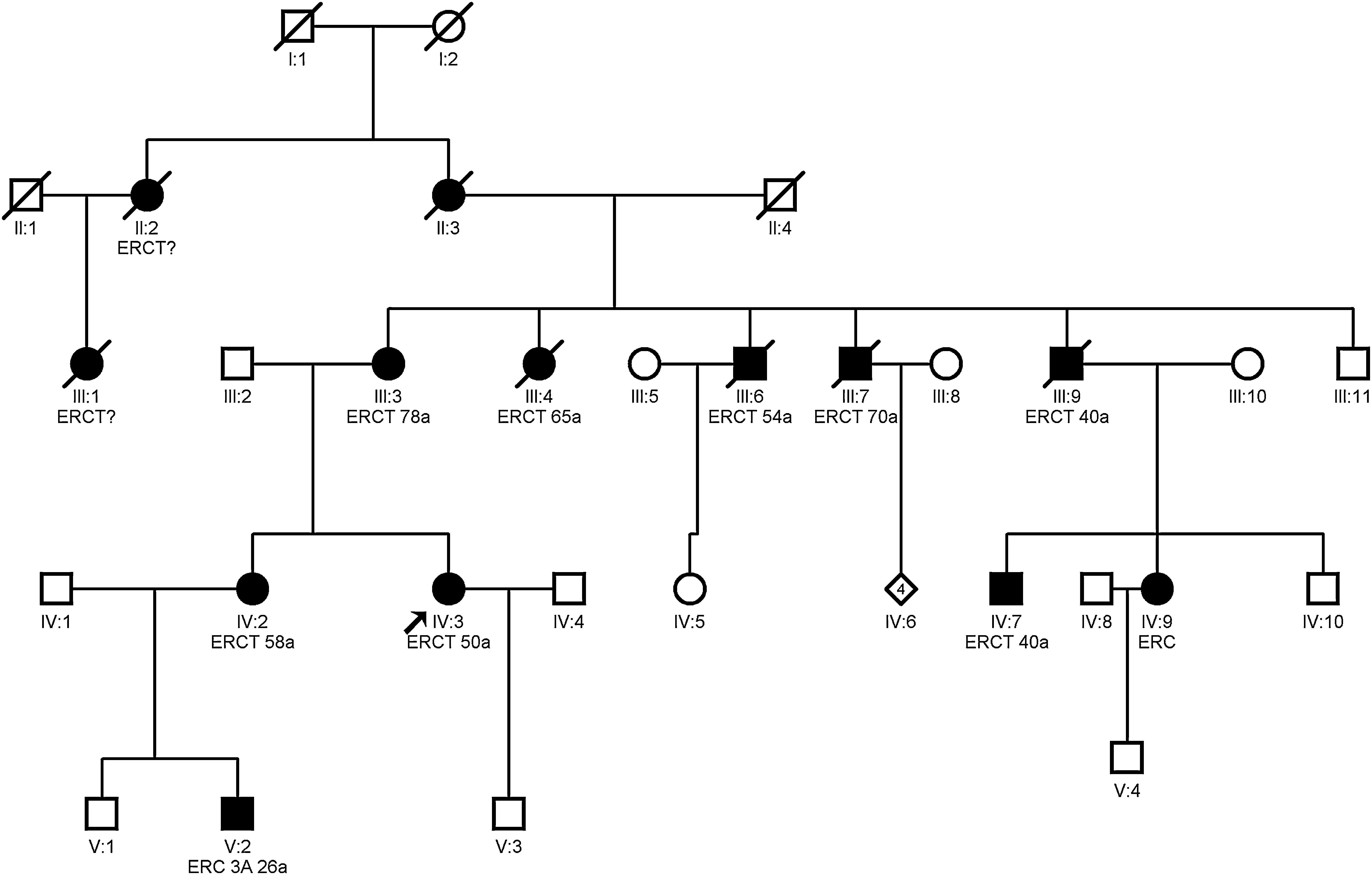
The MUC1 gene (chromosomal region 1q22) was identified as the cause of ADTKD in 2013,30 although linkage studies had identified the candidate locus on chromosome 1q21 in 1998.31 The first family affected was described in Cyprus in 2002.22 This gene, of 7 exons, codes for mucin-1, a highly glycosylated transmembrane protein largely expressed in the distal nephron. The function of mucins is to preserve the luminal epithelial barrier. The mutation consists of the duplication of a cytosine from a chain of 7 cytosines located in variable numbers (20–125) in a 60 base pairs repeate in tandem (VNTR). A change in the readingframe occurs (frameshift), resulting in an altered protein (MUC1-fs). It is not known why this abnormal gene product causes in the kidney tubulointerstitial fibrosis and it does not produce damage in another tissues where mucin-1 is also expressed (breast, lung, sebaceous and salivary glands).26
Clinical featuresPatients with mutation in the MUC1 gene do not present any extrarenal manifestation.13Fig. 3 shows the example of a family with ADTKD due to mutation in the MUC1 gene.
Autosomal dominant tubulointerstitial nephropathy caused by mutations in the HNF1B gene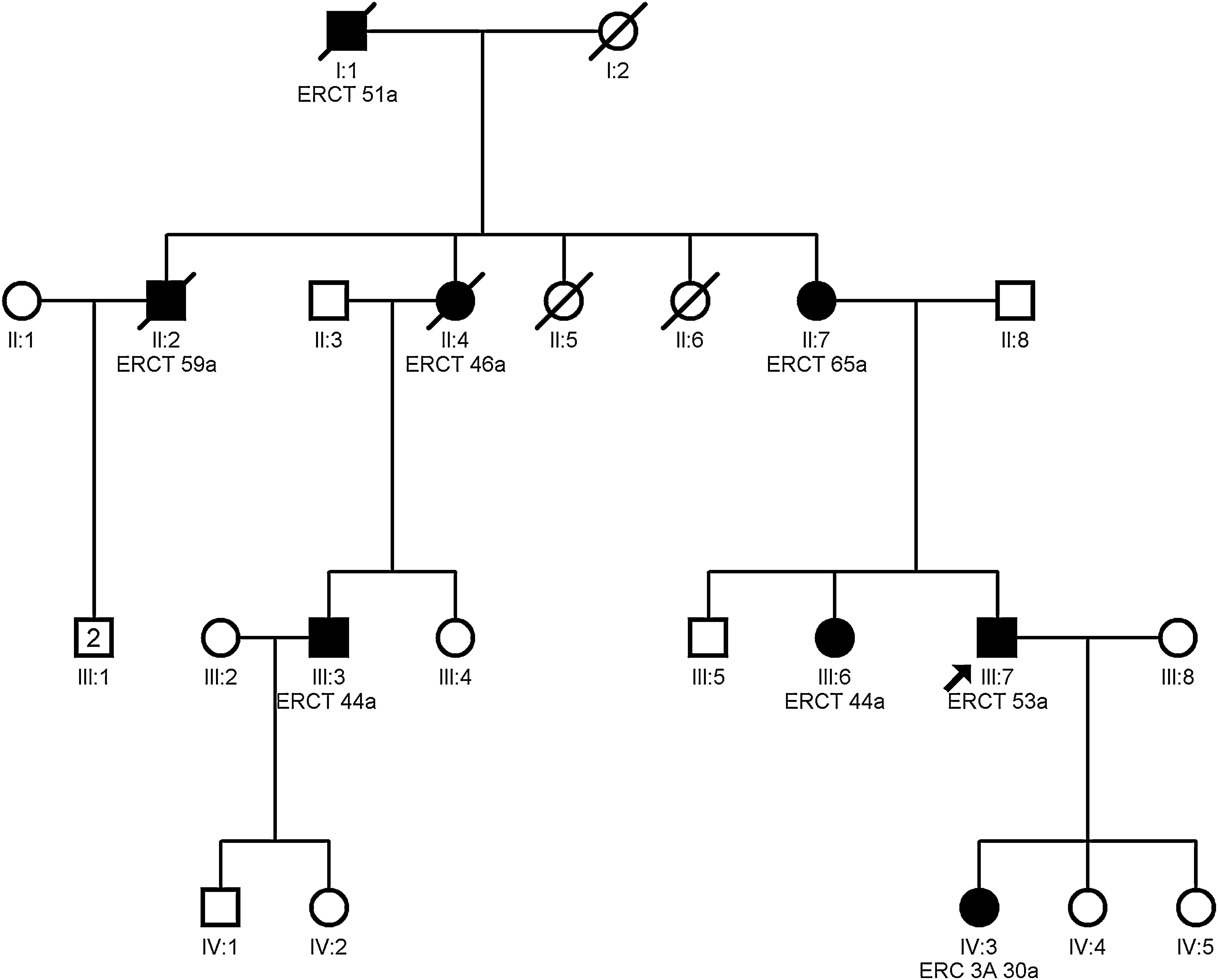
The HNF1B gene (OMIM 17q12 and 189907) codes for hepatic nuclear factor 1b (HNF1b), a transcription factor that regulates multiple genes expressed during the embryogenesis of different tissues (renal, hepatic, pancreatic or genital).32,33 The HNF1B gene in turn regulates the UMOD gene and genes involved in autosomal dominant polycystic kidney disease.34
Clinical featuresMutations of the HNF1B gene may cause multiple extrarenal abnormalities and only a minority have in tubulointerstitial nephropathy.35,36 Only those clinical forms with tubulointerstitial fibrosis as the main manifestation are considered as ADTKD.11,16,32 Early symptoms may occur in infancy, and even in the prenatal period.
Autosomal dominant tubulointerstitial nephropathy caused by mutations in the REN geneThe REN gene (OMIM 1q32.1 and 179820) encodes encodes preprorenin, which is subsequently proteolytically processed to prorenin and renin. Abnormal renin is accumulated in cells producing renin causing apoptosis.17 Renin is also expressed in the tubular cells of the distal nephron, interrelating with abnormal tubular expression of other ADTKD genes.37
Clinical featuresThe clinical features of the disease can be attributed to the low activation of the renin angiotensin aldosterone system. Renin and aldosterone levels are low with tendency to hyperkalemia and some degree of hypotension. Consequently, patients are at increased risk of renal failure in situations of volume depletion or use of NSAIDs.38,39 Anemia occurs early and it is disproportionate to the degree of renal failure; it may be seen in childhood, disappear in adolescence, and reappear in more advanced stages of kidney disease.17 To date, there are only 14 families identified with a mutation in the REN40 gene.
Criteria for suspected diagnosisThe suspicion criteria, established by the KDIGO guidelines of ADTKD,11 are described in Table 3, and Fig. 4 shows an algorithm for diagnostic orientation.
Criteria for the diagnosis of ADTKD.
| A. Diagnostic suspicion |
| • Family history compatible with an autosomal dominant inheritance pattern and chronic kidney disease with the characteristics mentioned above (see Table 2). |
| • In the absence of a family history that fulfills the previously indicated characteristics, histological demonstration by renal biopsy or extrarenal manifestations compatible with mutation in HNF1b or history of hyperuricemia or early gout. |
| B. Criteria to establish the diagnosis |
| • Family history compatible with a pattern of autosomal dominant inheritance and chronic kidney disease with the characteristics mentioned above (see Table 2) and compatible histological demonstration by renal biopsy in at least one affected family member (definitive diagnosis is possible only by Renal biopsy). |
| or |
| • Demonstration of a mutation in one of the 4 genes in the patient or at least in one member of the family. |
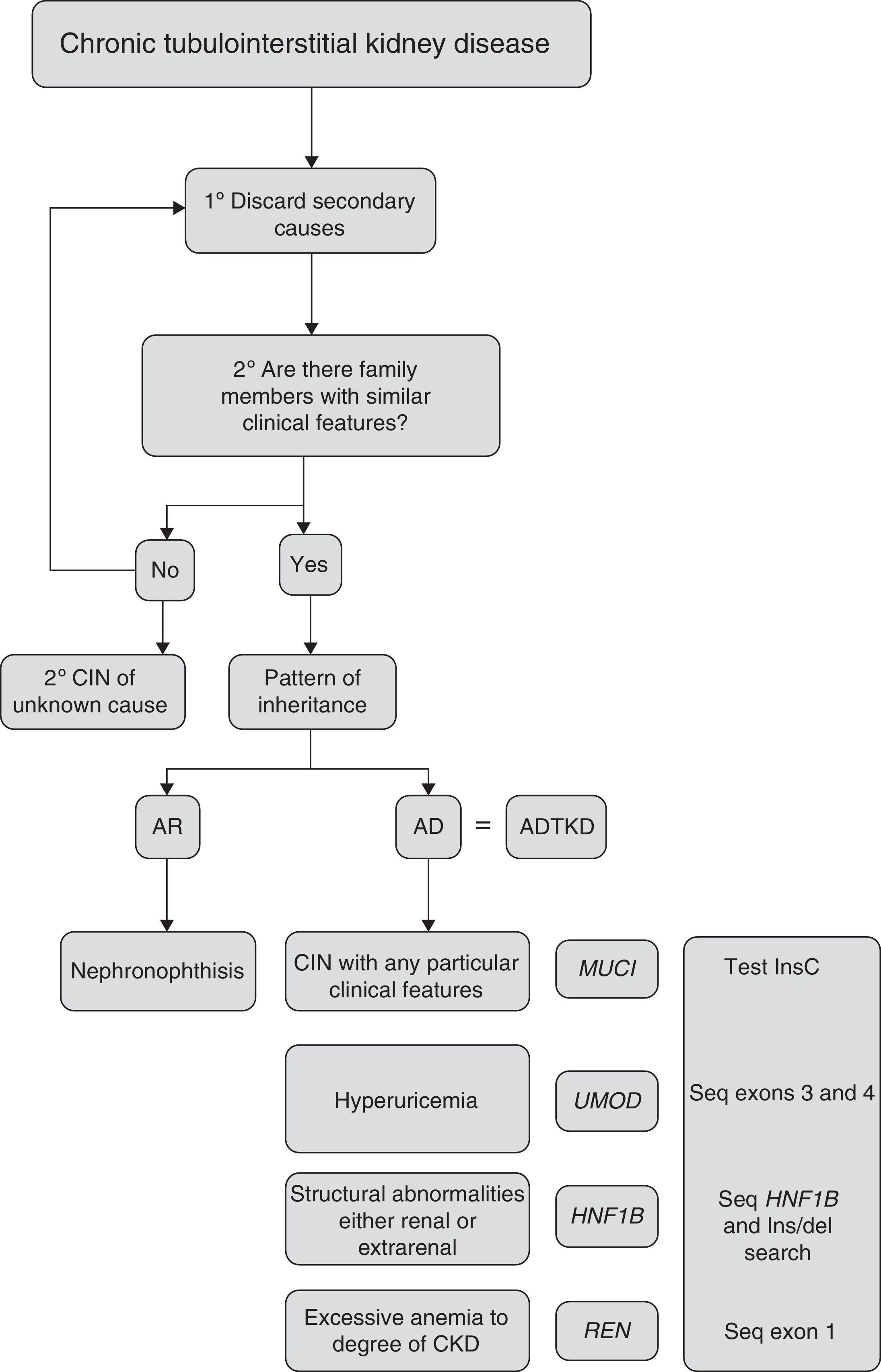
This diagnostic algorithm is proposed for the evaluation of patients with suspected chronic interstitial nephropathy. After clinical suspicion, it should be assessed if there is a family history of nephropathy with similar characteristics. Subsequently, based on the most outstanding clinical features, establish a diagnostic priority. The recommended genetic study is indicated. CIN: chronic interstitial nephropathy; AR: autosomal recessive; AD: autosomal dominant; ADTKD: autosomal dominant tubulointerstitial kidney disease.
If there is a positive family history with autosomal dominant inheritance pattern and there is a renal biopsy compatible with the disease in at least one family member, the rest of the family with suggestive clinical features does not usually need a biopsy.
ADTKD should also be considered in patients, with no family history, but with the previously mentioned characteristics. These may be considered de novo cases or with wrong diagnoses in other relatives. These cases should be considered as “ADTKD suspects” if histology is not compatible.11
The only method to confirm the diagnosis and to correctly classify the type of ADTKD is through Genetic analysis and rule out the disease in other family members. Not all the genes that may cause ADTKD have been identified therefore negative genetic result does not totally exclude the diagnosis. In addition, genetic diagnostic techniques do not have a sensitivity of 100%. The analysis of the UMOD, REN and HNF1B genes are well established,12 however the MUC1 gene is much more complex and the analysis is performed only in some specialized centers.30 Such a study cannot be performed simply by Sanger or massive sequencing, since the only mutation of MUC1 known so far is located in a tandem repeat region rich in guanine–cytosine (GC-rich 60-base VNTR). There are only few cases described with MUC1 mutation. The mutation has been identified in some elderly individuals without symptoms so there could be incomplete penetrance (asymptomatic mutation carriers).
Recommendations for the genetic studyDespite the fact that there is no available specific treatment for ADTKD, genetic analysis is recommended to make a precise diagnosis, to identify of the subtypes of mutations and to determine whether family members have the disease. Table 4 describes the situations in which genetic study is recommended.
Recommendations to perform the genetic study.
| • Confirm diagnosis in adults with suspected ADTKD |
| • Potential kidney donors of affected families |
| • In cases of preimplantation genetic diagnosis |
| • Adults at risk from a family with ADTKD with an identified mutation |
| • Pediatric patients with suspected REN mutation |
ADTKD: autosomal dominant tubulointerstitial kidney disease.
If the mutation of the disease is identified, patients should receive genetic counseling. The possibility of genetic study should also be offered to other members of the family. In general, it is not recommended to perform genetic studies in children, since there is no specific therapy anyway. The case is different for suspicion of mutation in the REN gene, since they could benefit from early treatment with EPO or fludrocortisone.40
Recommendations for management and follow-up- •
Genetic study is recommended in patients with suspected ADTKD.
- •
Genetic study of at risk-family members is recommended.
- •
In individual cases with symptoms suggesting chronic interstitial nephropathy, with no family history, the return of genetic studies is very low. Better think about other causes of kidney disease.
- •
It is recommended to control the risk factors (renal hypertension, diabetes mellitus, smoking, obesity) that may influence kidney damage and perform an annual analysis of renal function.
- •
In children at risk of mutation disease in UMOD or MUC1, the options for treatment are scarce. Children at risk for a mutation in HNF1B or REN should be referred to the pediatric nephrologist. In these cases they would benefit from an earlier management.
- •
NSAIDs should be avoided in all patients with ADTKD and especially those with mutations in the REN gene, as they are more susceptible to deteriorate renal function after NSAIDs.
- •
Salt restriction is not recommended in patients with genetic defects in UMOD and REN: it may aggravate hyperuricemia.
- •
It is not known whether the diet poor in purines is beneficial in patients with mutations in UMOD.
- •
In general, diuretics should be used with caution, as they may aggravate hyperuricemia and volume depletion.
- •
Patients with UMOD-associated disease who develop gout may suffer from new episodes. Gout can be prevented with allopurinol and, if it is not tolerated, an alternative is febuxostat once the first crisis has been resolved.
- •
There is no consensus on the benefits of renin–aldosterone blockade for prevention of renal disease progression. Losartan is not recommended since it increases uric acid excretion.
- •
In cases of REN gene mutation, patients usually require EPO early and also fludrocortisone to treat hypotension; however it should not be used in patients with deterioration of glomerular filtration, hypertension, hyperkalemia or edema.
- •
Renal transplantation is the treatment of choice in ESRD caused by ADTKD, since the disease does not recur in the graft.
- •
Renal-pancreatic double transplantation may be considered in patients with mutation in HNF1B and with diabetes.
Key concepts.
| • The term. ADTKD has recently been established to group these diseases of autosomal dominant inheritance. The concept of medullary cystic diseases is no longer applicable |
| • There are 4 genes described responsible for these diseases: UMOD, MUC1, HNF1B and REN. Other potential genes remain to be identified. There is a very important group of ADTKD families with unknown mutations |
| • ADTKD is a rare cause of hereditary kidney disease, with anodyne and heterogeneous presentation, with great inter- and intrafamily clinical variability. Although the penetrance is elevated, in some cases of slow progression with comorbidities or early deaths, the diagnosis may be difficult. Renal involvement is characterized by tubulointerstitial fibrosis and slow progression to end-stage renal disease |
| • Corticomedullary cysts are not always present so they are not longer a diagnostic criteria. Isolated cases of chronic interstitial nephropathy with no family history rarely are de novo cases; in these cases it is advisable to rule out other causes of CKD before requesting a genetic study. De novo mutations are more frequent in the HNF1b gene |
CKD: chronic kidney disease; ADTKD: autosomal dominant tubulointerstitial kidney disease.
The authors declare no conflict of interest.
The study has been supported by: ISCIII RETIC REDINREN RD16/0009 FEDER FUNDS, FIS PI15/01824, FIS PI13/01731.


AL DÍA

