




Presentamos un caso de inicio de nefropatía lúpica (NL) como glomerulonefritis fibrilar (GNF).
Mujer de origen rumano de 17 años que inició, en agosto del 2021, con eritema malar y gonalgia inflamatoria. Su médico de Atención Primaria objetivó función renal conservada, con ANA y anti-DNA positivos, microhematuria y albuminuria, por lo que se derivó a nuestra consulta. Sin fiebre, pérdida de peso, otras artralgias, artritis, uveítis, Raynaud ni aftas. Menstruaciones regulares. Sin obesidad. Sin otros antecedentes personales de interés. Su madre padecía hipertensión arterial, psoriasis e hipotiroidismo autoinmune. Se realizó una biopsia de piel sobre el eritema malar con resultado de lupus cutáneo agudo. Se completó estudio en el que destacaban: hemograma y bioquímica en rango de normalidad salvo proteínas totales, 5,6g/dl, y albúmina en plasma, 2,5g/dl. Orina elemental con proteinuria 30mg/dl, hematíes 3-10 por campo; CCr: 246ml/min/1,73 m2 y proteinuria: 2,4g/24h.
Autoinmunidad con ANA positivo patrón homogéneo AC1 420 título 1/160, anticuerpos anti-DNA nativo positivo (título 243,3). Ac. anti-DNA nativos (Crithidia luciliae) positivo. El resto, incluyendo anticuerpos antifosfolípidos y ß-2 glicoproteína 1, negativos. C3: 61mg/dl, C4: 9mg/dl. Perfil tiroideo con TSH normal y anticuerpos antimicrosómicos-TPO 107,60 UI/ml (valores normales: 0,0-60,0). Ecografía de cuello, normal. Serologías con inmunoglobulinas (Ig) G positivas e IgM negativas para VEB, rubéola, VHS tipo 6, varicela zóster. Quantiferon y serología negativos frente a leishmania, leptospira, Tripanosoma cruzi, Borrelia, Strongyloides, parvovirus, lúes, citomegalovirus, toxoplasma, VHB, VHC, VIH, Brucella y Bartonella.
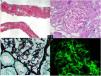
Se realizó biopsia renal (fig. 1) con resultado de 19 glomérulos por plano de corte, 1 con esclerosis global del ovillo capilar (5%). Los glomérulos conservados mantenían su tamaño y lobulación normales. Asas capilares finas y permeables. Sin depósitos en asas de alambre, seudotrombos hialinos, lesiones necrosantes, cariorrexis, neutrófilos ni hipercelularidad endo o extracapilar. Ligera expansión mesangial sin incremento de celularidad. En 2 glomérulos, y con distribución segmentaria, imágenes que sugieren proyecciones espiculares sin imágenes de vacuolización de la membrana basal (MB) ni claros depósitos subepiteliales en la inmunofluorescencia directa (IFD). Túbulo intersticio sin alteraciones significativas. Vasos de la pared muscular y arteriolas sin lesiones. Sin fibrosis intimal, hialinosis arteriolar ni material amiloide con el rojo congo. IFD: depósito granular mesangial y subendotelial en asa capilares de IgG (++) y C3 (+++), y mesangial de IgM (+) y C1q (++), con leve predominio de cadenas ligeras kappa frente a lambda. Diagnóstico de NL clase III. Score de actividad: 0/24. Score de cronicidad: 1/12.

Biopsia renal. Microscopia óptica. A) Parénquima cortical renal con arquitectura general conservada sin cambios crónicos significativos (tricrómico de Masson). B) Glomérulos sin expansión mesangial, lesiones proliferativas ni otras alteraciones morfológicas (PAS, 20×). C) Glomérulo con proyecciones espiculares («spikes») de la membrana basal de forma muy focal (plata, 40×). D) IFD: depósito de IgG y C3 granular mesangial y en asas capilares e IgM y C1q mesangial (IgG, 40×).
Se inició tratamiento con IECA, hidroxicloroquina, corticoides y micofenolato, y al cabo de 6 meses de seguimiento mantiene la función renal estable y la proteinuria ha disminuido a 0,65 g/24 h.
En este momento se reciben los resultados de la microscopia electrónica (fig. 2): 4 glomérulos de arquitectura general distorsionada. Podocitos con cambios vellocitarios de su citoplasma, sin depósitos intracitoplasmáticos. Pedicelos focalmente fusionados con afectación del 30% de la extensión pedicelar total y reordenamiento de los filamentos intermedios hacia el polo basal de la célula. MB de contornos irregulares, difusamente engrosada, espesor de la lámina densa entre 700 y 925nm (promedio de 863nm, VN: 300-400nm), depósitos difusos moderadamente electrodensos, organizados en forma de fibrillas cuyo diámetro oscila entre 10,60 y 35,65nm (promedio 18,32nm), engastados en la lámina densa y que ocasionan reacción espiculada de la MB glomerular, sin estar incrustados en la misma. Escasos depósitos subendoteliales. Endotelios de aspecto conservado, con fenestraciones patentes e inclusiones tubulorreticulares. Mesangios moderadamente ampliados a expensas de matriz y con depósitos electrodensos frecuentes de iguales características a los anteriormente descritos. Túbulos y vasos sin lesiones ultraestructurales. Sin depósitos en las MB tubulares ni vasculares. Ante estos hallazgos se realizó tinción inmunohistoquímica para DNAJB9 resultando positiva en patrón granular en la vertiente subepitelial de la MB, lo que otorga un diagnóstico de glomerulonefritis fibrilar.

Microscopia electrónica. A) Glomérulo con fusión pedicelar extensa (flechas negras). B) Presencia de inclusiones túbulo-reticulares en el endotelio. C) Depósitos moderadamente electrodensos de localización subepitelial (*) con reacción espiculada de la lámina densa (flechas blancas). D) Depósitos organizados en forma de fibrillas dispuestas de forma desorganizada. Microscopio electrónico de transmisión A y C: 5.000×, B: 8.000×, D: 1.2000×. LC: luz capilar; MBG: membrana basal glomerular; NCE: núcleo de la célula endotelial; P: podocito.
Ante la buena respuesta inicial al tratamiento inmunosupresor, este se mantiene a la espera de evolución.
La proteína DNA JB9 es una cochaperona que se sitúa en el retículo endoplásmico de muchas células, se libera con el estrés e interviene en el plegamiento de las proteínas. En la GNF1,2 con un mecanismo de depósitos de inmunocomplejos se han demostrado Ig con restricción para IgG4 e IgG1 lo que se explica porque la Ig sea la proteína que se deposita o porque se deposite una proteína DNA JB9 mal plegada a nivel glomerular y desencadene la reacción inflamatoria subsecuente. El mecanismo depende de la activación del complemento por la vía clásica (C4 positiva en tinciones de forma generalizada). En la serie más amplia publicada de GNF3 se describe un 23% de casos asociados a tumores que iniciaron desde 15 años antes a 10 después (mieloma múltiple, leucemia mielomonocítica crónica, tiroides, hepatocelular, pulmón, útero, próstata, colon, renal, melanoma), un 15% asociada a enfermedades autoinmunes sin especificar el porcentaje (lupus, Sjögren, enfermedad de Cröhn, Graves-Basedow, cirrosis biliar primaria, púrpura trombótica idiopática y espondilitis anquilosante), 3% con hepatitis C crónica, 20% diabetes, 9% cardiopatía isquémica crónica, 5% EPOC y un 17% con gammapatía monoclonal. Clínicamente cursó en un 71% de casos con HTA, 59% con edemas, 41% con proteinuria (media de 3,1 g/día), 62% hipoalbuminemia, 38% hipercolesterolemia, 62% síndrome nefrótico completo, 52% hematuria, 29% leucocituria y 2% hipocomplementemia.
La asociación GNF con NL4-10 se ha descrito de forma excepcional en la recidiva tras trasplante renal, en cuadros de NL sin lupus sistémico y en un caso de neurolupus a partir del cual se llega al diagnóstico de NL clase 4.
En una revisión de 223 biopsias renales de NL con depósitos electrodensos organizados11 se detectaron 3casos con depósitos tubulares y 2casos con depósitos de fibrillas, con las características estructurales de glomerulopatía inmunotactoide y GNF.
Con respecto al pronóstico, dado el bajo número de casos no hay datos concluyentes, aunque la GNF evoluciona a enfermedad renal terminal en un 50% de los casos a los 5 años. Por la misma razón, no hay evidencias con respecto al tratamiento, aunque se recomienda el uso de rituximab.
En nuestro caso, dado el escaso tiempo de evolución, solo podemos hablar de respuesta parcial a los 2 meses de tratamiento.
FinanciaciónEste artículo no tiene fuentes de financiación.



