





Diabetes, dyslipidemia, older age, gender, urinary tract infections, and recent antibiotic intake have been associated with a decrease in the urobiome richness and other fluctuations in this microbiome. Gut and blood microbiome have been reported to be altered in patients with chronic kidney disease (CKD), and specifically in peritoneal dialysis (PD) patients. Still, there are currently no studies describing the urogenital microbiome in CKD-PD patients. In this study we characterized the urobiome profile in 46 PD patients and analyzed its clinical and inflammatory parameters.
Materials and methodsMid-stream urine, fecal and blood samples were collected from 46 patients undergoing PD at Centro Hospitalar Universitário de São João (CHUSJ) in Porto, Portugal. Exclusion criteria were age under 18 years old, inability to give informed consent, history of infection in the last three months, and antibiotic intake in the last three months. The microbiome communities were analyzed by amplification and sequencing of the V3–V4 region of the bacterial 16S rRNA gene. Correlations with the patients’ clinical data and inflammatory profile were performed.
ResultsCKD-PD patients presented a unique urobiome profile dominated by Bacillota, Actinomycetota and Pseudomonadota and characterized by a lower Shannon diversity than fecal and blood microbiome. The taxonomic profiles of urogenital samples were organized in multiple subtypes dominated by populations of Lactobacillus, Staphylococcus, Streptococcus, Gardnerella, Prevotella, Escherichia-Shigella, being similar to other non-PD-CKD patients. Gender, sCD14, residual diuresis and history of peritonitis were significantly associated to variations in the urobiome. Although not reaching statistical significance, diabetes and the time on PD also showed association with particular taxonomic groups. Depletion of Gardnerella, Staphylococcus, Corynebacterium, Lactobacillus or Dermabacter populations correlated with CKD-PD patients with history of diabetes, history of peritonitis and altered levels of sCD14.
ConclusionsOur results highlight urogenital microbiome as a potential partner and/or marker in the overall health state of CKD-PD patients.
Diabetes, dislipemia, edad avanzada, género, infecciones del tracto urinario y toma reciente de antibióticos, entre otras, se han asociado a una disminución en la riqueza del urobioma y a otras fluctuaciones de dicho microbioma.Recientemente, se han descrito alteraciones en losmicrobiomas intestinal y en sangreen pacientes con enfermedad renal crónica (ERC) y, específicamente, en pacientes en diálisis peritoneal (DP).A pesar de ello, aún no existen estudios que describan el microbioma urogenital en pacientes en DP. En el presente trabajo, caracterizamos el urobioma en 46 pacientes en DP.
Pacientes y métodosSe recogieron muestras de orina (micción espontánea), heces y sangre de 46 pacientes en DP del Centro HospitalarUniversitário de São João en Oporto, Portugal. Los criterios de exclusión fueron edad menor a 18 años, incapacidad para entenderel consentimiento informado, e historia de infección y toma de antibióticos en los últimos 3 meses. Las comunidades microbiológicas fueron analizadas por amplificación y secuenciación de las regiones V3-V4 del 16S rRNA bacteriano. Se realizaron correlaciones con los datos clínicos y el perfil inflamatorio de los pacientes.
ResultadosLos pacientes en DP presentaron un urobioma único dominado por Bacillota, Actinomycetota yPseudomonadota, y caracterizado por una menor diversidad de Shannon que los microbiomas en sangre e intestinal. Los perfiles taxonómicos de las muestras urogenitales se organizaron en múltiples subtipos dominados por poblaciones de Lactobacillus, Staphylococcus, Streptococcus, Gardnerella, Prevotella, Escherichia-Shigella, siendo similar al descrito para otros pacientes con ERC no en DP.Género, factor sCD14, diuresis residual yantecedentes de peritonitis se asociaron de forma significativa a cambios en el urobioma. Diabetes y tiempo en DP también se asociaron con grupos taxonómicos específicos, aunque no de forma estadísticamente significativa. La deplección de Gardnerella, Staphylococcus, Corynebacterium, Lactobacillus o Dermabacterse correlacionó con antecedentes de diabetes, historia de peritonitis y niveles alterados de sCD14.
ConclusionesNuestros resultados subrayan la importancia del microbioma urogenital como potencial biomarcadordel estado de salud de nuestros pacientes en DP.
Chronic kidney disease (CKD) is a worldwide health problem carrying a high socioeconomic burden with elevated morbidity and mortality.1 CKD may progress to different stages resulting in a decrease of the ability of the kidneys to excrete liquids and toxic products in the urine. When the estimated glomerular filtration rate (eGFR) is lower than 15mL/min per 1.73m2, patients with CKD require renal replacement therapy including kidney transplantation, hemodialysis, and peritoneal dialysis (PD).2 In PD the peritoneum acts as a high vascularized dialysis membrane and allows the removal of water and solutes from the internal milieu. In comparison to hemodialysis, PD might enable a better quality of life for the patients, and can also preserve the residual renal function of the patients for a longer period.3 Therefore, CKD-PD patients may maintain residual diuresis for longer periods and at higher values than hemodialysis patients. Still, PD-related infections, specially peritonitis and catheter exit-site infections, represent the major problem of PD units, as these infections can be responsible for catheter loss, transfer to hemodialysis, prolonged hospitalization, and, in more severe cases, death of these patients.4
Gut microbiome has been shown to be altered in CKD-PD patients.5–7 These alterations, also known as gut dysbiosis, contribute to the chronic inflammatory state of these patients. Particularly in the gut, CKD-PD patients present a significantly decreased bacterial diversity and a gut microbiome dominated by urease containing indole- and p-cresyl-forming bacteria.8 Inversely, taxa linked to the production of probiotic butyrate, short-chain fatty acids (SCFAs), and carbohydrate fermentation appear to be markedly reduced.9 A reduction in SCFAs production may be linked to longer dialysis duration, higher peritoneal glucose exposure, and loss of residual renal function. A recent study also correlated PD-protein-energy wasting with a decrease in butyric acid-producing bacteria, namely Roseburia and Phascolarctobacterium, and with an increase of bacteria associated to intestinal permeability, inflammation, and nutritional imbalance, such as Escherichia.10Dorea, Clostridium, and SMB53 were reported to be less abundant in CKD-PD patients with peritonitis, suggesting that these bacteria may have anti-inflammatory roles.11 A recent study also proposed a decrease in the ratio Bacillota/Bacteroidota (formerly Firmicutes/Bacteroidetes) as a potential biomarker of Escherichia coli peritonitis in these patients.12 Beyond the gut, blood and peritoneum microbiomes were also recently described to be compromised in CKD-PD patients.7,13,14
Urine has been considered throughout the history a sterile fluid. However there is evidence showing that the urinary tract of healthy individuals without urinary infection is dominated by different kinds of viable microbes and the distribution pattern of these microorganisms may affect the urinary tract health.15–17 Urinary microbiome, or urobiome, is an emerging topic in health and disease, specifically in urinary tract disorders.15,17,18 Such differences could be observed in both bladder and voided urine.17 Urinary dysbiosis has been associated with urinary tract infections and incontinence,19 overactive bladder syndrome,20,21 neuropathic bladder,16,22 chronic prostatitis,23 nephrolithiasis,24–26 and urinary tract neoplasms,27 and other systemic factors such as diabetes, dyslipidemia, older age, gender, and recent antibiotic intake.28,29 For example, patients with kidney stone disease and hypertension had a unique microbiome profile and such changes may reflect disease progression and may be useful to monitor response to treatments.24 Also, some studies have observed a different urine microbiome composition in CKD patients when comparing with individuals with normal kidney function18 and a reduction of diversity associated to a decrease in renal function.30 Besides, some studies on the urinary microbiome of CKD-kidney transplant recipients (KTR) revealed an overall decrease in diversity and emergence of opportunistic pathogens promoting antibiotic resistance.31,32 Moreover, alteration in the urinary microbiome of KTR have been related to chronic allograft dysfunction and increased susceptibility to infection.20
There are still no studies describing the urogenital microbiome (by testing voided urine) in CKD-PD patients, possibly due to the loss of residual renal function and, consequently, their diuresis. Therefore, this study aimed to evaluate the urogenital microbiome of CKD-PD patients’ population (n=46) by throughput sequencing and compare it with their gut and circulating (blood) microbiomes. Also, we aimed to analyze the patients’ inflammatory markers.
Materials and methodsPatient selection and sample collectionThis cross-sectional observational study included 46 CKD patients undergoing PD at Centro Hospitalar Universitário de São João in Porto, Portugal. We invited all the prevalent patients from the PD unit that had a medical appointment at the hospital's outpatient clinic during the four months of recruitment period (September 2018 to January 2019), except those with exclusion criteria. The exclusion criteria for this study included age under 18 years old, inability to give informed consent, history of infection in the last three months, and antibiotic intake in the last three months. Clinical and demographic information was collected for each participant, regarding gender, age, CKD etiology, diabetes mellitus, dyslipidemia, obesity (defined as body mass index of 30kg/m2 and higher), and cardiovascular disease (peripheral vascular disease, ischemic cardiopathy, or cerebrovascular disease). The modality of PD (automated or manual), pharmacological treatment, and history of infection were also registered.
Blood samples were collected in the PD unit, as previously described.7 At the same day, the self-collected stool and mid-stream urine specimens were brought refrigerated by the patient within 4h or 48h after self-collection, for urine and stool samples, respectively. When stool was collected more than 4h before out-patients consultation, they were frozen. After aliquoted, the whole blood, urine, and stool samples were immediately frozen and stored at −80°C until microbiome analysis.
Routine clinical analyses were performed testing proteinuria, and serum levels of urea, albumin, hemoglobin, cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), triglycerides, phosphorus (P), calcium (Ca), calcium phosphate product, ferritin, B-type natriuretic peptide (BNP), parathyroid hormone (PTH), sedimentation velocity (SV), C-reactive protein (CRP), creatinine clearance (Ccreat), residual renal function, and Kt/V (urea). Kt/V (urea) is a parameter that measures adequacy to PD using urea weekly clearance normalized by urea estimated distribution volume. Tumor necrosis factor α (TNF-α), IL-1, IL-6, IL-10 were determined in plasma by Luminex Multiplex Assay (Millipore). ELISA kits were used to evaluate lipopolysaccharide-binding protein (LPS-BP, Cloud-clone Corp®), Toll-like receptor 4 (TLR4, Cloud-clone Corp®), and soluble CD14 (sCD14, Quantikine® ELISA).
Sample processing for microbiome analysisGenomic DNA was isolated in a strictly controlled environment at Vaiomer SAS (Labège, France). Total DNA was extracted from whole blood (100μL) and urine using a protocol carefully designed to minimize risks of contamination.33 Extraction of DNA from feces was done using QIAamp Fast DNA Stool Mini Kit (Qiagen). Negative controls (molecular grade water added in an empty tube, the same used for sample storage and PD solution) were used simultaneously with the tested samples. PCR amplification was performed using universal primers targeting the V3–V4 region of the bacterial 16S rRNA gene (340F-781R) to encompass 476-base pair amplicon. The usage of 2×300 paired-end MiSeq kit V3 was followed. Sample multiplexing and sequencing library generation were conducted, as previously described.33 qPCR was used to quantify the DNA concentration in the pool employing a 7900HT Fast Real-Time PCR System (Life Technologies) and KAPA Library Quantification Kits for Illumina Platform (Kapa Biosystems, Inc.). The final pool with a concentration between 5 and 20nM (after dilution) was used for sequencing in a MiSeq Illumina device, as previously suggested.13
16S rRNA gene sequencing analysisThe targeted gene regions were initially analyzed using the FROGS bioinformatics pipeline established by Vaiomer SAS (Labège, France).34 Some filters were applied as previously suggested13: (1) amplicons with a length <350 nt or a length >480 nt were removed; (2) amplicons without the two PCR primers were removed (10% of mismatches were authorized); (3) amplicons with at least one ambiguous nucleotides (‘N’) were removed; (4) operational taxonomic units (OTUs) identified as chimera (with search v1.9.5) in all samples in which they were presented were removed; (5) OTU with an abundance lower than 0.005% of the whole dataset abundance were removed, and (6) OTU with a strong similarity (coverage and identity ≥80%) with the phiX (library used as a control for Illumina sequencing runs) were removed. OTU were obtained via single-linkage clustering and the taxonomic assignment performed by Blast+ v2.2.30+ with the RDP v11.4 databank.
Statistics and data analysisPrimer v7 (PRIMER-e, Auckland, New Zealand) was used for the calculation of diversity indices, as well as, non-metric multidimensional scaling (NMDS) and principal coordinate analyses (PCO), and other multivariate analyses, mainly analysis of similarities (ANOSIM) and permutational multivariate analysis of variance (PERMANOVA), used to test the significance of β-diversity. The percentage of each OTU per sample was used for these analyses, followed by squared root transformed data, resemblance matrices of similarity data types using Bray–Curtis similarities, adding dummy value and testing 4999 permutations. The reads in each sample were initially converted into percentage values according to the total number of sequences in the sample to eliminate the effect of the final number of reads.35 Post hoc analyses were done in STAMP 2.1.336 and the statistical tests used for multiple groups were one-way analysis of variance (ANOVA), Tukey–Kramer (0.95) and Eta-squared for effect size, while two groups were analyzed using Welch's t-test (two-sided, Welch's inverted for confidence interval method).
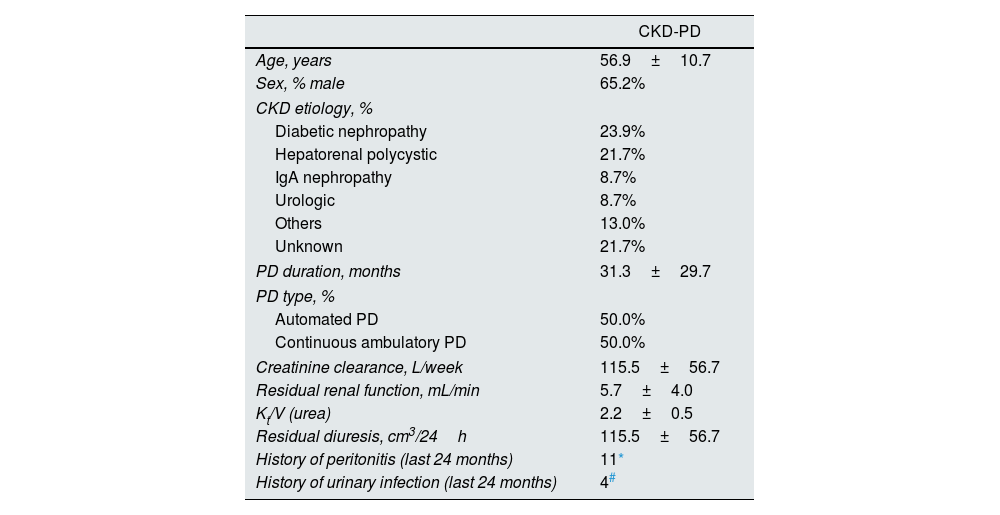
ResultsUrobiome characterization in CKD-PD patientsThe detailed characterization of patients on peritoneal dialysis included in this study is shown in Tables 1 and 2.
Characterization of chronic kidney disease patients on peritoneal dialysis (CKD-PD).
| CKD-PD | |
|---|---|
| Age, years | 56.9±10.7 |
| Sex, % male | 65.2% |
| CKD etiology, % | |
| Diabetic nephropathy | 23.9% |
| Hepatorenal polycystic | 21.7% |
| IgA nephropathy | 8.7% |
| Urologic | 8.7% |
| Others | 13.0% |
| Unknown | 21.7% |
| PD duration, months | 31.3±29.7 |
| PD type, % | |
| Automated PD | 50.0% |
| Continuous ambulatory PD | 50.0% |
| Creatinine clearance, L/week | 115.5±56.7 |
| Residual renal function, mL/min | 5.7±4.0 |
| Kt/V (urea) | 2.2±0.5 |
| Residual diuresis, cm3/24h | 115.5±56.7 |
| History of peritonitis (last 24 months) | 11* |
| History of urinary infection (last 24 months) | 4# |
Cases caused by Staphylococcus epidermidis (n=2), Streptococcus salivarius (n=2), Rhizobium radiobacter, Klebsiella oxytoca, Streptococcus mitis, Pantoea spp., Serratia marcescens, and unidentified agent (n=2). Patients with infections in the last 3 months were excluded from this study and are not included here.
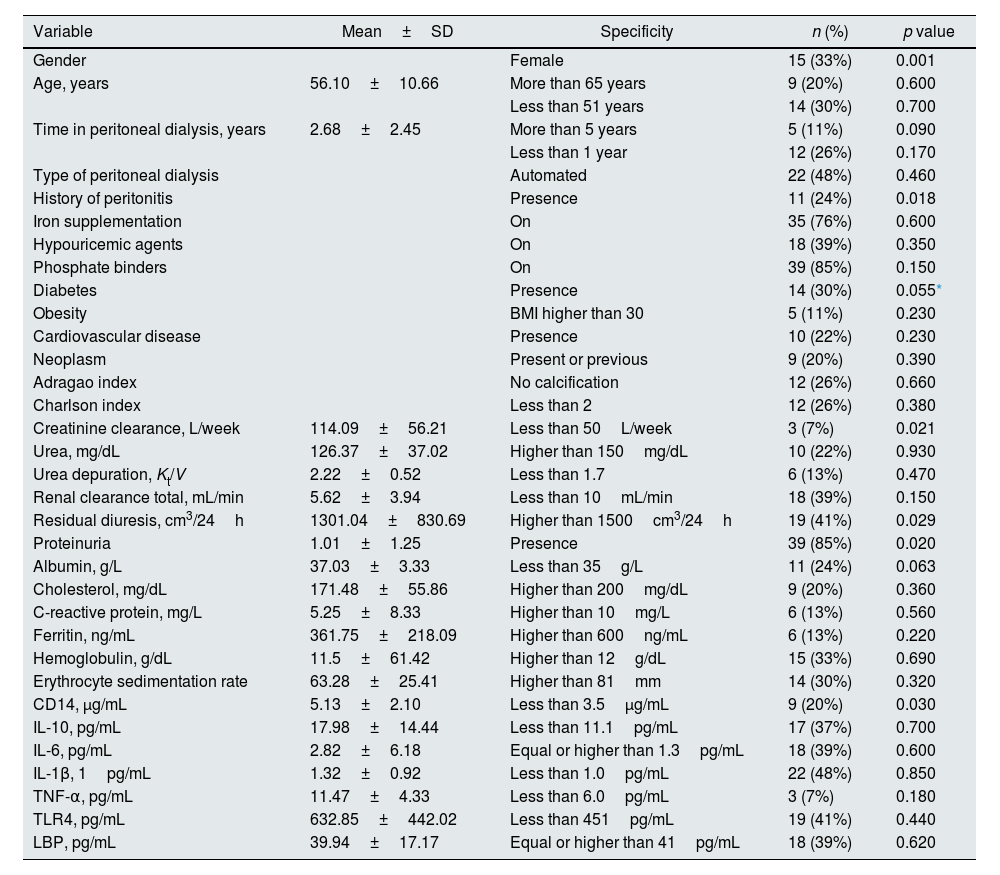
Demographic and clinical characterization of PD-CKD patients and the association of these variables with urinary microbiome. Results expressed in mean and standard deviation (mean±SD) for continuous variables and number of patients (n) and prevalence (%) for categoric variables; for statistical analysis, continuous variables were transformed in categoric variables.
| Variable | Mean±SD | Specificity | n (%) | p value |
|---|---|---|---|---|
| Gender | Female | 15 (33%) | 0.001 | |
| Age, years | 56.10±10.66 | More than 65 years | 9 (20%) | 0.600 |
| Less than 51 years | 14 (30%) | 0.700 | ||
| Time in peritoneal dialysis, years | 2.68±2.45 | More than 5 years | 5 (11%) | 0.090 |
| Less than 1 year | 12 (26%) | 0.170 | ||
| Type of peritoneal dialysis | Automated | 22 (48%) | 0.460 | |
| History of peritonitis | Presence | 11 (24%) | 0.018 | |
| Iron supplementation | On | 35 (76%) | 0.600 | |
| Hypouricemic agents | On | 18 (39%) | 0.350 | |
| Phosphate binders | On | 39 (85%) | 0.150 | |
| Diabetes | Presence | 14 (30%) | 0.055* | |
| Obesity | BMI higher than 30 | 5 (11%) | 0.230 | |
| Cardiovascular disease | Presence | 10 (22%) | 0.230 | |
| Neoplasm | Present or previous | 9 (20%) | 0.390 | |
| Adragao index | No calcification | 12 (26%) | 0.660 | |
| Charlson index | Less than 2 | 12 (26%) | 0.380 | |
| Creatinine clearance, L/week | 114.09±56.21 | Less than 50L/week | 3 (7%) | 0.021 |
| Urea, mg/dL | 126.37±37.02 | Higher than 150mg/dL | 10 (22%) | 0.930 |
| Urea depuration, Kt/V | 2.22±0.52 | Less than 1.7 | 6 (13%) | 0.470 |
| Renal clearance total, mL/min | 5.62±3.94 | Less than 10mL/min | 18 (39%) | 0.150 |
| Residual diuresis, cm3/24h | 1301.04±830.69 | Higher than 1500cm3/24h | 19 (41%) | 0.029 |
| Proteinuria | 1.01±1.25 | Presence | 39 (85%) | 0.020 |
| Albumin, g/L | 37.03±3.33 | Less than 35g/L | 11 (24%) | 0.063 |
| Cholesterol, mg/dL | 171.48±55.86 | Higher than 200mg/dL | 9 (20%) | 0.360 |
| C-reactive protein, mg/L | 5.25±8.33 | Higher than 10mg/L | 6 (13%) | 0.560 |
| Ferritin, ng/mL | 361.75±218.09 | Higher than 600ng/mL | 6 (13%) | 0.220 |
| Hemoglobulin, g/dL | 11.5±61.42 | Higher than 12g/dL | 15 (33%) | 0.690 |
| Erythrocyte sedimentation rate | 63.28±25.41 | Higher than 81mm | 14 (30%) | 0.320 |
| CD14, μg/mL | 5.13±2.10 | Less than 3.5μg/mL | 9 (20%) | 0.030 |
| IL-10, pg/mL | 17.98±14.44 | Less than 11.1pg/mL | 17 (37%) | 0.700 |
| IL-6, pg/mL | 2.82±6.18 | Equal or higher than 1.3pg/mL | 18 (39%) | 0.600 |
| IL-1β, 1pg/mL | 1.32±0.92 | Less than 1.0pg/mL | 22 (48%) | 0.850 |
| TNF-α, pg/mL | 11.47±4.33 | Less than 6.0pg/mL | 3 (7%) | 0.180 |
| TLR4, pg/mL | 632.85±442.02 | Less than 451pg/mL | 19 (41%) | 0.440 |
| LBP, pg/mL | 39.94±17.17 | Equal or higher than 41pg/mL | 18 (39%) | 0.620 |
The bacterial microbiomes evaluated in this study resulted in a total of 1127 OTUs obtained from the analysis of urine, gut, and blood samples from 46 CKD-PD patients. A set of 583 OTUs were found in urine samples, while gut samples showed 542 OTUs, and blood samples 514 OTUs. Regarding the Shannon diversity, urine showed the lower diversity in comparison to the gut and blood diversity: the median values was 2.29 for urine (ranging from 1.19 to 4.14), 3.41 for gut (2.39–4.09), and 2.66 for blood (1.79–3.09), with significance difference among all groups of samples (p<0.001). Regarding to other diversity indexes, urine samples showed a richness of 11.3 (range 2.0–44.7) and an evenness of 0.5 (range 0.1–0.8). The taxonomic profiles among the three habitats were distinct (Fig. 1A). Urine samples were dominated by Bacillota, similarly to gut samples, but also comprised Actinomycetota (formerly Actinobacteria), and Pseudomonadota (formerly Proteobacteria). At family level, Streptococcaceae, Enterobacteriaceae, Lactobacillaceae, Family XI, and Bifidobacteriaceae were the most frequent groups in urine samples (Fig. 1B). The microbiome of the tested samples was significantly different comparing urine, gut, and blood (p=0.001, Fig. 2).
 phylum and (B) family levels.")

The taxonomic profiles allowed to organize the urogenital samples in multiple subtypes, comprising samples dominated by Lactobacillus, Staphylococcus, Streptococcus, Gardnerella, Prevotella, Escherichia-Shigella, or others (Fig. 3). The value of these subtypes is complex, but it is not related to active urinary infections (because patients with active infections were excluded). Nevertheless, all urogenital samples of Lactobacillus subtype were linked to female patients, while the Staphylococcus and Anaerococcus dominated samples were exclusively found in male patients.
, namely Lactobacillus, Staphylococcus, Streptococcus, Gardnerella, Prevotella, Escherichia-Shigella, or other subtypes (no specific genus dominates this subset of samples).")
Sample subtypes observed among the urobiome of patients on peritoneal dialysis. Each subtype is dominated by a specific taxonomic group (genus), namely Lactobacillus, Staphylococcus, Streptococcus, Gardnerella, Prevotella, Escherichia-Shigella, or other subtypes (no specific genus dominates this subset of samples).
In this study Gardnerella OTUs were exclusively found in urogenital samples (in 20 out of 46 samples). Dermabacter and Atopobium OTUs were rarely observed in gut and blood samples (in 1 and 2 samples, respectively), but more frequently found in urine (6 samples were positive for Dermabacter and 13 samples for Atopobium). There were other taxonomic groups only found in urogenital samples (see supplementary Table S1). When urine OTUs were compared with OTUs in gut and blood samples, some similarities were also observed. There were three OTUs (identified as Pseudomonas and Stenotrophomonas) common for all patients included in this study, and two OTUs (identified as Pelomonas and Escherichia-Shigella) presented in more than 90% of the patients. Interestingly, there were some OTUs found in multiple samples from a single patient (e.g. urine and blood). A set of 54 OTUs were common to multiple samples from a single patient, being 39 of these OTUs (72%) found in urogenital and fecal samples. Only 9 OTUs (17%) were simultaneously observed in urogenital and blood samples (17%), and 6 OTUs (11%) were observed in fecal and blood samples. Similarities were also high when the analysis was extended to OTUs present in more than one patient, suggesting a high interplay between taxa in urogenital and gut microbiomes.
Association between taxonomic groups and demographic and clinical factorsDemographic and clinical variables of CKD-PD patients were studied to find correlations with the urogenital microbiome (Table 2), being the largest significance found for gender (p=0.001), particularly of Lactobacillus linked to females and other families, such as Staphylococcus and Anaerococcus, to males (Fig. 4). Fig. 4 shows the taxonomic groups significantly affected by the separation of urogenital samples in two groups according to different clinical variables. Patients with more than 3.5μg/mL soluble CD14 levels showed increased levels of Lactobacilllus, Dermabacter, and Gardnerella in urine microbiome (p=0.03) when compared with patients with equal or lower levels of sCD14 (Fig. 4). Lower relative abundance of Atopobium, Dermabacter, and Gardnerella were found in diabetic CKD-PD patients (p=0.055; p=0.04 by removing two outgroup samples as shown in supplementary Fig. S1). Residual diuresis lower than or equal to 1500mL/24h, proteinuria, and Ccreat lower than 50L/week were also associated to differences in the PD urobiome, while longer periods on PD (>5 years) and albumin did not significantly affect the urobiome (Table 2); multiple taxa were associated to these microbiome differences (Fig. 4 and supplementary Fig. S1). Curiously, patients with residual diuresis lower than or equal to 1500mL/24h also showed a lower Shannon diversity (median value of 1.66) than patients with normal values (median value of 2.41).
 considering the clinical and demographic factors associated to urobiome; all the remaining taxa not displayed in the figures are similar in both groups of patients – the figure displays only the differences. Gender, sCD14, residual diuresis, history of peritonitis, proteinuria and creatinine clearance were significantly associated to urobiome profiles (p<0.05; see Table 2 with values for the analysis of similarities (ANOSIM) and confirmed by permutational multivariate analysis of variance (PERMANOVA). Post hoc analyses were done in STAMP 2.1.3.")
Post hoc analyses describing prevalence differences of multiple taxonomic groups (and p values associated to each taxa) considering the clinical and demographic factors associated to urobiome; all the remaining taxa not displayed in the figures are similar in both groups of patients – the figure displays only the differences. Gender, sCD14, residual diuresis, history of peritonitis, proteinuria and creatinine clearance were significantly associated to urobiome profiles (p<0.05; see Table 2 with values for the analysis of similarities (ANOSIM) and confirmed by permutational multivariate analysis of variance (PERMANOVA). Post hoc analyses were done in STAMP 2.1.3.
History of peritonitis was also associated with differences in the urogenital microbiome, being Gardnerella, Staphylococcus, and Corynebacterium decreased in such patients (Fig. 4). Previous peritonitis was reported in 11 patients, being caused by Staphylococcus epidermidis (n=2), Streptococcus salivarius (n=2), Rhizobium radiobacter, Klebsiella oxytoca, Streptococcus mitis, Pantoea spp., Serratia marcescens, while no agents were identified in 4 cases. Also, there were 4 cases of reported urinary infections (occurring more than three months before samples collection) among these patients, being caused by E. coli (n=2), Streptococcus haemolyticus (n=1) and one agent was unidentified. Neither the history of peritonitis, nor previous urinary infection was associated to particular subtypes described above and in Fig. 3.
In our work, we also measured markers of intestinal translocation (endotoxins, LPS-binding protein, TLR4, and sCD14), inflammatory parameters (C-reactive protein, ferritin, sedimentation velocity, IL-1β, IL-6, TNF-α, and the anti-inflammatory IL-10), and other routine laboratory parameters (such as urea, proteinuria, albumin, hemoglobin, cholesterol and its different fractions, triglycerides, calcium, parathormone, BNP), but no statistically significant differences were found (Table 2). No difference was observed on the urogenital microbiome of patients depending on their PD modality (automated or manual).
DiscussionUrobiome relevance in CKD-PD and other CKD patientsThe existence of an urobiome has been recognized since 201037 and multiple studies have described its diversity and complexity.16,17,28,30,38 The urinary microbiome has not been previously described in CKD-PD patients; these patients preserve residual renal function. In the present study, the urogenital microbiome of CKD-PD patients showed a lower biodiversity (Shannon index) compared to gut and blood microbiomes, as described for other non-PD-CKD patients.25 The gut and blood microbiome profiles were recently published for CKD-PD patients.7 We found that the urogenital microbiome of our patients was distinct from the gut and blood microbiome and presented some exclusive taxa such as Gardnerella.
In our population, the urobiome was dominated by Bacillota, Actinomycetota and Pseudomonadota, specifically by the families Streptococcaceae, Enterobacteriaceae, Lactobacillaceae, and Bifidobacteriaceae, as previously described for CKD patients stages 3–5 that were not on dialysis.30 The urine subtypes described in this set of samples agreed with the previously described subtypes,30,39 being dominated by the genera Lactobacillus, Staphylococcus, Streptococcus, Gardnerella, Prevotella, and Escherichia-Shigella, suggesting that such subtypes may be transversal to distinct groups of CKD patients30 and may have a role in CKD pathophysiology. Interestingly, in the study of Kramer et al.30 the urobiome diversity decreased as kidney function worsened and similar observation was seen in this study for CKD-PD patients with lower values of residual diuresis.
Demographic and clinical factors and urobiomeIt has been recognized the impact of the urinary microbiome on recurrent urinary infections among women.40 Patients with history of infections and antibiotic intake in the last three months were excluded, so no conclusions can be drawn about that in this study. Nevertheless, previous history of peritonitis was described as an important factor associated to changes in the urobiome in this study, being Gardnerella, Staphylococcus, and Corynebacterium decreased in these patients. It is possible that peritonitis promotes a microbial translocation to the bladder, and this may alter the patient urobiome persistently even after successful treatment. Catheter exit-site infections were not associated to urogenital microbiome differences in our PD patients.
Additionally, some clinical conditions, such as diabetes, dyslipidemia, older age, or gender,28 have been associated with a decrease in the urogenital microbiome richness and other fluctuations in the urogenital microbiome. In line with previous reports, we found that gender and diabetes were associated to differences in CKD-PD urobiome, while age did not alter this microbiome. Regarding the gender, Lactobacilllus was previously described in the urobiome of healthy and CKD women,30 corroborating our findings. Moreover, we found Staphylococcus increased in the urobiome of CKD-PD men, also as previously described.30
Residual diuresis, proteinuria, and creatinine clearance (Ccreat) were associated to changes in the urobiome of our CKD-PD. We observed higher levels of Corynebacterium and Staphylococcus in patients with residual diuresis ≤1500mL/24h. The genera Staphylococcus and Corynebacterium, among others, have been shown to be increased in patients with urinary tract infection.41 Interestingly, we observed that urobiome diversity decreased with lower values of residual diuresis. Kramer et al.,30 in a previous study in CKD patients not on dialysis, also showed that urobiome diversity decreased as kidney function worsened. The reduction in the residual diuresis is an indicator of loss of residual renal function and a risk factor for poor outcomes and prognosis in PD patients.42 So, the changes in the urobiome that accompany the loss of residual renal function in CKD-PD patients should be further explored. Some species of Corynebacterium have a potent ability to metabolize urea and some studies relate an increase of this bacterium to urinary calculi.26,43 We also observed that proteinuria and Ccreat lower than 50L/week were associated to different taxonomic groups in the CKD-PD urobiome. Still, such findings result from a low number of samples analyzed and should be further confirmed.
The levels of inflammatory parameters and intermediate size molecules clearance may be related to specific taxonomic groups frequently present in urine,23,40 but confirmatory studies supporting these associations are still lacking. Interestingly, in our study CKD-PD patients with higher sCD4 levels presented an increase in Lactobacillus, Dermabacter, and Gardnerella. CD14 is a human monocyte differentiation antigen that acts as a pattern recognition receptor by binding to pathogen-associated molecular patterns such as lipopolysaccharides (LPS), working as toll-like receptor (TLR) co-receptor for the detection of infections.44 Particularly in CKD, higher levels of sCD14 have been associated to vascular calcification, cardiovascular disease and have been proposed as a predictor of all-cause mortality.7,45 sCD14 role in CKD-associated dysbiosis should be explored in future studies.
Origin of urogenital microorganismsIn this study, the depletion of OTUs of Atopobium or Dermabacter, and Gardnerella in our CKD-PD patients was significantly associated with history of diabetes, history of peritonitis, and lower levels of sCD14. Gardnerella has been identified in the urobiome of patients with history of high blood pressure24 and has also been described in the vagina microbiome of healthy women and women with bacterial vaginosis.39,46 The increase of Gardnerella in the vagina and the urogenital microbiomes has been associated to disease. In our CKD-PD patients, the reduction of Gardnerella was associated to diabetes and history of peritonitis, but the specific role of this bacterium is still unclear in these patients. Gardnerella has been shown to be depleted in the urobiome of diabetic patients compared to healthy controls and was not found in a diabetes plus dyslipidemia cohort comparing with other cohorts (diabetes only, diabetes plus hypertension, and diabetes plus hypertension and dyslipidemia).47
CKD-PD patients may present a different quantitative and qualitative microbial profile in gut, blood, and urine when compared to healthy individuals. Changes in the gut and blood microbiomes of CKD-PD patients have been previously reported.7 But there are no previous studies describing the urobiome of CKD-PD patients. In this study, we have described for the first time the urobiome of CKD-PD patients. Results showed shared OTUs between the gut, the blood, and the urine microbiomes. These similarities can be influenced by the uremic environment that promotes the disruption of intestinal tight junctions, and the translocation of gut taxonomic groups and toxins into the blood and other body fluids.15 Nonetheless, this is a unicentric, cross-sectional study. For this reason, future studies should explore unrecognized mobility pathways of the human microbiome through different body habitats, and its role in infections and systemic inflammation in CKD-PD patients.
ConclusionCKD-PD patients presented a unique urogenital microbiome with lower biodiversity than gut and blood microbiomes. History of peritonitis and relevant clinical biomarkers of CKD were associated to variations in the urobiome. Depletion of certain Gardnerella, Staphylococcus, Corynebacterium, Lactobacillus, or Dermabacter populations were observed in CKD-PD patients with history of diabetes, history of peritonitis and altered levels of sCD14. Further, multicentric studies will allow the better understanding of the mobility pathways of the human microbiome and the clarification of the impact of urobiome on CKD-PD patients outcomes, namely on the risk of peritonitis, loss of residual renal function, and recurrent urinary tract infections.
Ethical approvalThe study was approved by the local Ethics Committee (approval reference 200/18) in accordance with the 1964 Helsinki declaration and its later amendments. All participants agreed to participate in the study after receiving detailed information on the study objectives and protocol, and the written informed consent was obtained from all patients.
Data availabilityThe data supporting reported results can be found at NCBI under BioProject: PRJNA819486.
FundingR.A. was supported by Individual Call to Scientific Employment Stimulus – Second Edition (grant number CEECIND/01070/2018).
Competing interestsAll authors declare no conflict of interest.
The following are the supplementary data to this article:



