











Contrast agents can directly or indirectly induce renal tubular ischemia and hypoxic damage. Given that cobalt chloride (CoCl2) can protect renal tubules, the protective effect and potential mechanism of action of CoCl2 on contrast-induced nephropathy (CIN) warrant investigation.
MethodsA CIN mouse model was established to determine the protective effect of CoCl2 on renal injury in vivo. Then, TMT-based proteomics was performed to determine the differentially expressed proteins (DEPs), following which, enrichment analyses of gene ontology and the KEGG pathway were performed. In vitro, a CIN model was constructed with renal tubular epithelial cells (HK-2) to determine the effect of CoCl2 on potential targets and the role of the key protein identified from the in vivo experiments.
ResultsCoCl2 treatment decreased the levels of BUN and serum creatinine (sCr), while increasing the levels of urea and creatinine (Cr) in the urine of mice after CIN injury. Damage to the renal tubules in the CoCl2 treatment group was significantly less than in the CIN model group. We identified 79 DEPs after treating the in vivo model with CoCl2, and frequently observed ferroptosis-related GO and KEGG pathway terms. Of these, Hp (haptoglobin) was selected and found to have a strong renoprotective effect, even though its expression level in kidney tissue decreased after CoCl2 treatment. In HK-2 cells, overexpression of Hp reduced the ferroptosis caused by erastin, while knocking down Hp negated the attenuation effect of CoCl2 on HK-2 cell ferroptosis.
ConclusionCoCl2 attenuated kidney damage in the CIN model, and this effect was associated with the decrease in ferroptosis mediated by Hp.
Los agentes de contraste pueden inducir isquemia tubular renal y daño hipóxico de manera directa o indirecta. Dado que el cloruro de cobalto (CoCl2) puede proteger los túbulos renales, el efecto protector y el mecanismo de acción potencial de CoCl2 en la nefropatía inducida por contraste (NIC) merecen ser investigados.
MétodosSe estableció un modelo de NIC en ratones para determinar el efecto protector de CoCl2 en la nefropatía in vivo. Seguidamente, se realizó un análisis proteómico por TMT para determinar las proteínas diferencialmente expresadas (DEP) y, a continuación, un análisis de enriquecimiento de ontología genética y vía KEGG. In vitro, se construyó un modelo NIC en células epiteliales de túbulos renales (HK-2) para determinar el efecto de CoCl2 en los objetivos potenciales y el rol de la proteína clave identificada en los experimentos in vivo.
ResultadosEl tratamiento con CoCl2 redujo los niveles de BUN y de creatinina sérica e incrementó, a la vez, los de urea y creatinina en la orina de los ratones, tras la lesión NIC. El daño a los túbulos renales en el grupo de tratamiento con CoCl2 fue significativamente menor que en el grupo de modelo NIC. Identificamos 79 DEP tras el tratamiento en el modelo in vivo con CoCl2 y observamos con frecuencia ontología genética relacionada con ferroptosis y términos de vías KEGG. De ellos, se seleccionó la haptoglobina (Hp) y se encontró que tenía un fuerte efecto renoprotector, aun cuando su nivel de expresión en el tejido renal se redujo tras el tratamiento con CoCl2. En las células HK-2, la sobreexpresión de Hp redujo la ferroptosis causada por erastina, a pesar de que el descenso de Hp negó el efecto atenuador de CoCl2 en la ferroptosis de las células HK-2.
ConclusiónEl CoCl2 atenuó el daño renal en el modelo NIC y se asoció este efecto al descenso de ferroptosis mediada por Hp.
The number of patients with cardiovascular and chronic kidney diseases has increased, partly due to a rise in the elderly population. Consequently, there has been an uptick in the use of contrast agents for clinical diagnosis and treatment. A study showed that contrast-induced nephropathy (CIN) is a leading cause of acute kidney injury in hospitalized patients.1 CIN is characterized by an increase in serum creatinine levels of ≥0.5mg/dL (≥44μmol/L) or by ≥25% from the baseline value within 48h of contrast medium (CM) exposure, provided other causes of acute kidney injury are excluded.2 Risk factors for CIN encompass renal insufficiency and other clinical risk factors, such as advanced age, a low body mass index (BMI), myocardial infarction, cardiac insufficiency, inflammation, anemia, and bleeding.3 CIN prolongs hospital stays and increases patient mortality. Various risk factors can contribute to a high incidence of CIN among patients.4 The pathogenesis of CIN is multifaceted, with contributors, including the high osmotic pressure and viscosity of the CM, intrarenal hemodynamic disruptions, oxidative stress, and inflammatory reactions.5–7 Direct toxic effects of the CM on vascular endothelial cells, like apoptosis, a surge in intracellular calcium ion concentrations, and DNA fragmentation triggered by ROS, also significantly damage renal tubules.8
In the absence of detailed knowledge about the mechanisms at play, no definitive CIN treatment exists. Preventative measures, such as hydration therapy and drug prophylaxis, are routinely applied clinically to mitigate CIN incidence. Sodium bicarbonate hydration, normal saline hydration, and a combination of the two are prevalent hydration therapies. These hydration techniques can enhance blood flow, hastening CM excretion and thus diminishing its toxic impact on renal tubules.9 Nicorandil can curtail ROS accumulation in mitochondria, amplify renal tubular blood flow, and shield the kidneys from ischemic damage post-CM administration.10 Hypoxic preconditioning can heighten cell adaptability to hypoxic stress, considerably diminishing or even sidestepping the cell damage inflicted by hypoxia.11 Based on this insight, one study employed cobalt chloride (CoCl2) as a HIF-1α activator in a subtotal nephrectomy mouse model and discovered that this treatment reduced apoptosis in renal tubular cells, elevated renal tubular cell proliferation, and spurred angiogenesis.12 Another investigation revealed that treating HK-2 cells with CoCl2 tempered cell inflammation and curbed ROS buildup, thereby safeguarding the renal tubules.13 It remains to be ascertained whether CoCl2 can alleviate ischemic renal damage instigated by CIN and guard renal tubular cells. It is also crucial to probe the molecular and functional mechanisms steering this potential effect.
To discern the effects and the mechanism of action of CoCl2 on CIN, we devised a CIN mouse model. Post-CoCl2 administration, we gauged renal function indicators, conducted H&E staining to evaluate renal tubular harm, and executed immunofluorescence tests to gauge cell death rates in the renal epithelia. Subsequently, we pinpointed key target proteins using quantitative proteomics and bioinformatics tools. Both Western blotting and qPCR analyses were deployed to confirm the TMT findings. We also formulated an erastin injury model alongside a CIN HK-2 model to elucidate the interplay between the target protein and the renal protection proffered by CoCl2.
Materials and methodsAnimals and treatmentsMale BALB/c mice (3–4 weeks old) were purchased from Slack Laboratory Animal Co., Ltd. (Shanghai, China). All mice were housed at 22°C with 30% relative humidity under a 12-h/12-h light/dark cycle and were provided with food and water ad libitum. The experiment was approved and supervised by the Animal Protection and Use Agency Committee of Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine (approval number: XHEC-F-2021-069). All animal procedures adhered to the Laboratory Animal Guidelines for Ethical Review of Animal Welfare (GB/T 35892-2018). After a one-week adaptive feeding period, the mice were randomly divided into three groups (n=10 per group): the normal group, the CIN model group, and the CoCl2 treatment group. A model of contrast-induced nephropathy (CIN) was established using the method described in another study.14 Briefly, CIN was induced via the tail vein. As depicted in Fig. 1A, the administered drug was a hypotonic non-ionic contrast agent (iohexol) at a single dose of 1600mg iodine/kg, following the first injection of CoCl2 hexahydrate (Sigma, St. Louis, MO, USA), which was dissolved in distilled water and injected subcutaneously twice a day at a dose of 5mg/kg. The interval between CoCl2 administrations was 12h, and each group was treated for 72h. After each CoCl2 treatment, blood, urine, and kidney tissue samples were collected from each group of mice for analysis.

Time points in the animal experiment and kidney injury indicators. (A) Time points and experimental procedure. (B–E) Serum blood urea nitrogen (BUN), serum creatinine (sCr), urinary urea, and urinary creatinine after CoCl2 administration; * indicates model vs. normal, # indicates CoCl2vs. model; n=6; # p<0.05; **,## p<0.01.
ELISA test kits were utilized to assess the level of kidney injury indicators expression, which included blood urea nitrogen (BUN, ab83362, Abcam), serum creatinine (sCr, ab65340), urinary urea (ab93362), and urinary creatinine (ab65340). The ELISA plates were refrigerated overnight. To generate a standard curve, 100μL of the appropriate standard was mixed with 100μL of assay buffer in an ELISA plate well. After mixing, 100μL of the solution was transferred from one well to the next, repeated five times. Then, 90μL of assay buffer and 10μL of the diluted sample were added to the testing well. Next, 50μL of the diluted antibody solution was added to the corresponding wells. The plates were sealed and shaken for 3h. The blocking solution was discarded, and 400μL of wash buffer was added to each well. After a 1-min wash, the washing solution was removed, and this step was repeated four times. Subsequently, 100μL of the substrate solution was added to each detection well and incubated at room temperature for 15min. A stop reaction solution (100μL) was added to end the reaction, changing the color from blue to yellow. The OD value of each well's sample was measured at 450nm using a microplate reader. The concentration of the detection index for each sample was calculated based on the standard curve.
Hematoxylin and eosin (H&E) staining and PAS (periodic acid-Schiff) stainingInitially, kidney tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned into 5μm slices. The sections were deparaffinized and rehydrated through a gradient series of xylene and alcohol. They were stained with Harris’ hematoxylin solution for 3–8min and rinsed with double-distilled water, followed by eosin solution staining for 1–3min. The sections were then dehydrated through a gradient series of alcohols and xylene, dried, and sealed with neutral gum. Under a microscope, these sections were evaluated, and representative images were captured and analyzed. The sections also underwent PAS staining using standard protocols. Once stained, sections were observed under a light microscope, and kidney injuries were assessed and scored based on the proportion of cortical tubular necrosis: 0=normal, 1=1–10%, 2=11–25%, 3=26–45%, 4=46–75%, and 5=76–100%.15 The scoring was conducted in a blinded manner, and nine representative views were used for each sample (three biological replicates were taken from each group).
ProteomicsKidney tissue samples from each group were pulverized into powder in liquid nitrogen. Proteins were subsequently extracted using the SDT lysis method, comprising 4% (w/v) SDS, 100mM Tris/HCl pH 7.6, and 0.1M DTT. The BCA kit facilitated protein quantification. An adequate quantity of protein from each sample was trypsinized via the filter-aided proteome preparation (FASP) method. Lyophilized peptides were redissolved in a dissolution buffer (40μL) and quantified (OD280). For each group, 100μg of peptides were labeled with a tandem mass tag (TMT) using a commercial kit from Thermo Fisher (Waltham, USA). Once labeled peptides from all groups were combined, they were categorized using AKTA Purifier 100. The Easy nLC HPLC liquid system, operating at a nanoliter flow rate, further separated each fraction. Post-chromatographic separation, the Q-Exactive system (Thermo Fisher) was deployed for mass spectrometry analysis of samples. In primary mass spectrometry, the same peptide segment in different samples labeled with any TMT reagent displays an identical mass-to-charge ratio. In secondary mass spectrometry, the cleavable bond breaks, releasing the TMT reporter ion, producing 10 TMT reporter ion peaks in the mass spectrometer's low mass region. These intensities represent the relative expression of the peptide across different samples. Proteome Discoverer 1.4 16 facilitated protein identification and quantitative analysis.16 Proteins with an average ratio change of ≥1.2 or <0.83, and a p-value less than 0.05 were flagged as differentially expressed proteins (DEPs). A hierarchical clustering algorithm organized the differentially expressed proteins, and data was illustrated as a heatmap.
Bioinformatics analysisOmicsbean (http://www.omicsbean.cn/) annotated the suite of target proteins. This annotation included sequence alignment (Blast), GO entry extraction (Mapping), GO annotation (Annotation), and InterProScan supplementary annotation (Annotation Augmentation). DAVID Bioinformatics Resources v6.8 (https://david.ncifcrf.gov/) conducted the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Using the STRING (http://string-db.org/) database, both direct and indirect interactions among target proteins were discerned and visualized via Cytoscape, facilitating an interaction network analysis.
Cell culture and treatmentHuman kidney 2 (HK-2) cells were procured from the Cell Resource Center of the Institute of Basic Medicine, Chinese Academy of Medical Sciences. HK-2 cells thrived in DMEM enriched with 10% FBS, 100U/mL penicillin, and 100μg/mL streptomycin in a 37°C incubator. Cells were treated according to the requirements of the experimental groups. The normal group underwent routine cultivation. The model group received a supplement of 100mg/mL serum-free medium containing iohexol, and its osmotic pressure was fine-tuned to 424mOsm/L, establishing an in vitro model of contrast-induced acute kidney injury (CI-AKI). Stemming from the model group, the CoCl2 group received a 150μM CoCl2 dose. The Hp knockdown and Hp-overexpression groups were exposed to respective viral vectors for cell transfection. Upon treatment completion, cells were harvested for subsequent experiments.
TransfectionThe Hp shRNA, manufactured by GenePharm (Shanghai, China), was cloned into the pShuttle-H1 adenovirus plasmids. Additionally, the Hp coding sequence was synthesized and inserted into pShuttle-CMV adenovirus shuttle plasmids (Agilent, Beijing, China). Following the co-transfection of the blank pAdEasy-1 adenovirus skeleton plasmid (Addgene Headquarters, Watertown, MA, USA) with either pShuttle-H1-sh-Hp or pShuttle-CMV-Hp into HEK 293T cells using Lipofectamine 2000 (Invitrogen), a high-titer adenovirus vector was produced. After transfection, adenovirus plasmids generated through recombination were collected after 48h and then introduced into HK-2 cells. Negative controls were established using cells that were transduced with either the blank pShuttle-CMV plasmid (vector) or pShuttle-H1-nonspecific scramble shRNA plasmid (sh-NC). After 6h of transfection, the medium was replaced with fresh basal medium, and the culture was continued for 48h. Transfection efficiency, cell viability, and iron concentration were then determined via Western blotting assays.
Cell viability assayCell viability was measured using a CCK-8 kit (Beyotime, Shanghai, China) according to the manufacturer's instructions. Briefly, cells were seeded in a 96-well plate at a density of 2.0×103/well. After each group of cells underwent the specific treatment, 10μL of the CCK-8 reagent was added and incubated at 37°C for 2h. The absorbance of each sample was then evaluated using a microplate reader at 450nm (Tecan, Switzerland).
Iron quantificationInitially, cells were seeded at a density of 5×106 cells per plate, and each group was processed as required. Then, an iron detection kit (ab83366, Abcam) was used following the manufacturer's protocol. The cells were rinsed with ice-chilled PBS and homogenized (on ice) using an iron assay buffer that was five times the volume of the cell solution. The resultant mixture was then centrifuged at 13,000×g and 4°C for 10min to remove any insoluble material. The supernatant was collected, and each sample was treated with an iron reducer, mixed, and left undisturbed at room temperature for 30min. Each sample was treated with 100μL of the iron probe and mixed thoroughly either by pipetting or using a horizontal shaker. The reaction was allowed to progress for 60min at room temperature, with the plate kept in the dark. A microplate reader was used to determine the intracellular ion concentration at 593nm.
Analysis of the production of reactive oxygen species (ROS)After the cells were treated with CoCl2 in the absence or presence of iohexol, the superoxide indicator dihydroethidium (5μM; DHE, Invitrogen) was added to the culture medium. The cells were then incubated at 37°C for 1h. Nuclei were stained with DAPI. Fluorescence was observed under a fluorescence microscope (Revolve, Echo, San Diego, California, USA) and qualitatively analyzed. The excitation wavelength was 485nm, and the emission wavelength was 527nm.
Determination of ferroptosis by measuring lipid peroxidesTo visualize lipid ROS, cells were treated with 2μmol/L of the C11-BODIPY581/591 probe (Molecular Probes Inc.) and incubated in the dark at 37°C for 30min. Subsequently, DAPI (G1012; Servicebio) was used to stain the cell nuclei. Afterward, the cells were washed and analyzed using Liperfluo, and the fluorescence intensity of C11-BODIPY581/591 was determined.
RNA isolation and real-time PCRRNA was extracted from each group of tissues using an RNA extraction kit (Qiagen). Reverse transcription and quantitative PCR were performed following the instructions provided with the TAKARA (Dalian, China) reverse transcription and RT-PCR kit. The primers were as follows: Hp, 5′-AAACTCCCCGAATGTGAGGC-3′ (F), 5′-TCCATAGAGCCACCGATGATG-3′ (R); GAPDH, 5′-AGCTTCGGCACATATTTCATCTG-3′ (F), and 5′-CGTTCACTCCCATGACAAACA-3′ (R). The data were normalized to GAPDH and calculated using the 2−ΔΔCt method.
Western blotting analysisWestern blotting analysis was performed to detect the expression of the Hp and GAPDH proteins in kidney tissues and in HK-2 cells. The tissues or cells were routinely lysed to extract proteins. The BCA protein quantification kit (Thermo, USA) was used to quantify the extracted proteins. Conventional electrophoresis, membrane transfer, and antigen blocking were performed. Primary antibodies (dilutions: Hp (1:1000, EPR22856-212) and GAPDH (1:1000, ab8245)) were incubated with tissue sections overnight at 4°C. The following day, the sections were washed and incubated with a secondary antibody. The Tianneng imaging agent (Tianneng, Shanghai, China) was used to visualize the protein bands. All experiments were repeated in triplicate.
Immunofluorescence stainingImmunofluorescence double staining was performed to stain specific proteins expressed in kidney tissues from each group of mice, including E-cadherin and Hp. After the routine processes and before incubating with antibodies, the sections were placed in a 10% BSA humidified box and sealed in an incubator at 37°C for 30min. The sections were then incubated overnight at 4°C with diluted primary antibodies for E-cadherin (1:200, ab76055) and Hp (1:200). The next day, the slides were washed thrice with PBS (5min per wash). Then, the cy3 or FITC-labeled fluorescent secondary antibodies (that matched the primary antibody) were added to the sections and incubated for 2h. An anti-fluorescence quencher was added to the sections. Finally, the images were analyzed under a fluorescence microscope.
Statistical analysisAll experiments requiring statistical analysis were repeated in triplicate. Data are presented as the mean±standard errors. Kruskal–Wallis one-way analysis of variance (ANOVA) was performed using SPSS software (version 13.0, SPSS, USA). All differences among and between groups were considered statistically significant at p<0.05.
ResultsCoCl2 decreased renal function indicators after CINAfter administering cobalt chloride, we tested the levels of BUN, sCr, urinary urea, and urinary Cr in each group of mice. Following the addition of the contrast agent, the levels of BUN and sCr increased significantly, persisting until 72h post-operation. The levels of BUN and sCr in the CoCl2 group were significantly lower than those in the CIN group after 24h of modeling and remained so for 72h (Fig. 1B–E). The levels of urinary urea and urinary Cr were significantly lower in the CIN group but increased significantly after CoCl2 treatment.
CoCl2 attenuated the renal tubular damage caused by CINWe performed H&E staining on kidney tissues from each group. The stained samples showed prominent damage to the renal tubules in the model group, which included erythrocyte stasis in the renal tubules or corpuscles (red arrow), shedding of epithelial cells (green arrow), changes in the basement membrane (black arrow), and flattening of renal tubular cells due to tubular dilation (orange arrows). Renal injury was considerably less pronounced in the CoCl2 group (Fig. 2A). PAS staining yielded similar results. Tubule injury, characterized by cellular infiltration (red arrow) and dilated tubules with brush border loss (green arrows), epithelial flattening, and intratubular debris (black arrows), was evident in the contrast-induced model. However, CoCl2 treatment ameliorated this injury. Consistent changes in the injury score among groups also indicated the renoprotective effects of CoCl2 (Fig. 2B). We co-stained renal tubular cells with E-cadherin and TUNEL to assess cell death and observed that the number of TUNEL-positive renal tubular cells increased significantly in the model group. Yet, after CoCl2 treatment, the number of positive cells decreased significantly (Fig. 3).

Histopathological analysis after treatment. (A) H&E staining depicting kidney features. Model group exhibited erythrocyte stasis (red arrow), epithelial cell shedding (green arrow), bare basement membrane (black arrow), and renal tubular cell flattening due to dilation (orange arrows). CoCl2 group showed lesser damage than CIN group. (B) Periodic acid-Schiff staining: magnification, 400×. * indicateds model vs. normal, # indicates CoCl2 vs. model; n = 9; **,## p < 0.01.

To elucidate the mechanism underlying the action of CoCl2 on CIN, protein extracts from the kidneys of mice treated with or without CoCl2 were analyzed by TMT. After merging duplicated data, 5455 proteins were identified, with 79 being differentially expressed proteins (DEPs) between the model and CoCl2 groups (Fig. 4A and Supplementary Table S1). Through GO and KEGG pathway enrichment analyses, we discerned that these 79 proteins were primarily enriched in the homeostasis of several cell types (biological process, BP), the basal part of the cell (cell component, CC), and hemoglobin (Hb) binding (molecular function); including protein digestion and absorption, the ECM-receptor pathway, the PPAR signaling pathway, and steroid biosynthesis. Upon reviewing the literature, we noticed that some terms were associated with ferroptosis, which are enclosed in dotted lines (Fig. 4B and C). We then identified proteins that also changed differentially after iohexol induction, identifying 28 such proteins (Fig. 4D). Except for A2NW55, the remaining 27 proteins were all annotated: Krt78, Iigp1, Krt1, Col4a5, Hbb-b2, Col6a1, Acmsd, Slc43a1, Hbbt1, Arhgap12, Slc4a1, Cyp2e1, Bpgm, Alas1, Sptb, Cyp2d22, Adgrg1, Prss1, Pla2 g7, Flot2, Stfa2, Cd74, Nsmce1, Hmgcs2, Cyp4a14, Hp, and Gda. These proteins underwent GO and KEGG pathway enrichment analyses. The results revealed some terms associated with ferroptosis (enclosed in dashed boxes; Fig. 4E and F). After conducting the GO and KEGG pathway analyses for the 27 overlapping proteins, the terms related to ferroptosis were highlighted in a heatmap (Fig. 4G). We observed that five proteins (Cyp2d22, Pla2 g7, Sqle, Hp, and Cyp4a14) increased post-contrast induction and decreased after CoCl2 treatment, whereas the others did not follow this pattern. Therefore, we further analyzed them, noting that Sqle is a regulator of ferroptosis17 and Hp is also associated with it,18–20 but no published study has shown the relationship between other proteins and ferroptosis. Considering that Sqle plays a role in ferroptosis,17 and as Hp has a relatively uncertain function in ferroptosis, we selected Hp for further investigation, although our results showed that Hp did not have a high degree of connectivity in the PPI network of the 79 DEPs or the 27 overlapping DEPs (Supplementary Fig. S1).

Differentially expressed proteins (DEPs) and their GO and KEGG pathway analyses. (A) Heatmap of 79 DEPs (model vs. CoCl2 treatment) across all groups. (B and C) GO (B) and KEGG pathway (C) analyses of 79 DEPs. (D) Venn diagram of proteins overlapping between normal vs. model and model vs. CoCl2 comparisons. (E and F) GO (E) and KEGG pathway (F) analyses of 79 overlapped DEPs. (G) Proteins enriched in GO or KEGG pathways related to ferroptosis were selected; their expression among three groups is shown in the heatmap. Terms outlined with dotted lines were associated with ferroptosis based on published data.
To validate the results of the proteomics analysis, we conducted Western blotting and qPCR analyses to assess the expression of Hp's protein and mRNA in kidney tissues from the experimental mice. Our findings indicated that the protein level of Hp was notably higher in the model group than in the normal group. However, after CoCl2 administration, Hp's protein levels decreased (Fig. 5A). The mRNA level changes mirrored those observed in the protein levels (Fig. 5B). Dual fluorescent labeling was used to determine the expression level of Hp in renal tubular cells, aligning with the results from the TMT data (Fig. 5C). Based on these outcomes (Fig. 4), we deduced that Hp was intricately linked with ferroptosis. We then investigated cell viability, iron concentration, and ROS content in renal tissues to confirm whether CIN and CoCl2 are associated with ferroptosis. Our research established that cell viability decreased while iron concentration and ROS content increased in the renal tissues of CIN-afflicted mice. However, CoCl2 treatment partially reversed these effects, implying a potential relationship between CIN, CoCl2 treatment, and ferroptosis.

Western blotting, qPCR, and IF analyses of Hp from mouse kidney tissues. (A) Proteins tested thrice and normalized to GAPDH levels for quantitative analysis. (B) Quantitative analysis of Hp mRNA expression. IF co-stained four proteins and E-cadherin in renal tubular epithelial cells (C, magnification, 200×). (D–F) Cell viability, iron concentration, and ROS content estimated for ferroptosis levels. Each value represents mean±SD; * indicates model vs. normal; # indicates CoCl2vs. model; n=6; *,# p<0.05; **,## p<0.01.
HK-2 cells were utilized, along with Hp-interference and overexpression adenoviruses, to investigate the correlation between Hp and ferroptosis, as well as the potential role of Hp in the anti-CIN treatment with CoCl2. The efficiency of knockdown and/or overexpression strategies was assessed at both the transcriptional and translational levels following adenovirus infection (Fig. 6A and Fig. S2A–B). In vivo findings indicated a possible positive association between Hp and ferroptosis. However, when Hp was overexpressed in vitro, cell viability increased, a change that remained largely unaffected by the ferroptosis inhibitor ferrostatin-1 (Fer-1), which targets the 15LOX-PEBP1 complex and lipid peroxidation (Fig. S2C). To corroborate our observations, we assessed Fe concentration, ROS levels, and the expression of ferroptosis markers GPX4 (glutathione peroxidase 4) and SLC7A11 (solute carrier family 7 member 11). The results demonstrated that Hp inhibited ferroptosis without inducing iron overload, and this inhibition remained unaffected by Fer-1 (Fig. S2D–F). Consequently, Hp was deemed a ferroptosis suppressor. Subsequent experiments involved the use of erastin to induce ferroptosis in HK-2 cells. Erastin treatment reduced cell viability, an effect counteracted by Hp overexpression but exacerbated by Hp knockdown (Fig. 6B). Post-erastin treatment, cellular iron and ROS levels in HK-2 cells significantly increased. Notably, Hp overexpression markedly reduced iron and ROS levels, while Hp knockdown resulted in their significant elevation (Fig. 6C and D). Hp knockdown increased erastin-induced lipid oxidation, as indicated by increased green fluorescence intensity (reflecting levels of oxidized C11 BODIPY 581/591), while Hp overexpression diminished fluorescence intensity (Fig. 6E). Furthermore, protein levels of two typical ferroptosis biomarkers, Gpx4 and Slc7a11, decreased upon Hp knockdown and increased upon Hp overexpression following erastin treatment (Fig. 6F).

CoCl2's protective effect against CIN by regulating ferroptosis via Hp targeting. (A) Western blotting and qPCR to detect adenovirus transfection efficiency. Erastin-induced ferroptosis and Hp-interference/overexpression adenoviruses were transfected. (B–D) Cell viability, iron concentration, and ROS content estimated for ferroptosis level. (E) Ferroptosis analysis using C11-BODIPY581/591 fluorescence. (F) Western blotting of ferroptosis biomarkers GPX4 and SLC7A11 after erastin treatment with Hp overexpression or silencing. (G–I) Iohexol treated HK-2 cells to create CIN cellular model; CoCl2 or Hp-interference/overexpression adenoviruses applied. Cell viability, iron concentration, and ROS content measured for ferroptosis levels. (J) Ferroptosis analysis using C11-BODIPY581/591 fluorescence. (K) Western blotting of ferroptosis biomarkers Gpx4 and Slc7a11. Each value represents mean±SD; *p<0.05 and **p<0.01.
For creating an in vitro cellular model of CIN, HK-2 cells were treated with iohexol. CoCl2, in conjunction with Hp-interference or overexpression adenoviruses, was then employed to investigate whether CoCl2 can counteract CIN by modulating ferroptosis via Hp. The cellular activity of the CIN model group significantly decreased, while iron concentration and ROS content markedly increased. Following administration of CoCl2 or Hp overexpression, cell viability substantially increased, while iron concentration and ROS content significantly decreased compared to the CIN group (Fig. 6G–I). Lipid oxidation levels increased in the CIN group; however, after CoCl2 treatment or Hp overexpression, fluorescence intensity notably decreased (Fig. 6J). Changes in Gpx4 and Slc7a11 protein levels exhibited inverse patterns (Fig. 6K), thus, supporting the aforementioned findings (Fig. 6G–J). Conversely, Hp knockdown exhibited opposing effects on the above-mentioned ferroptosis indicators in the CIN in vitro model treated with CoCl2 (Fig. 6G–K).
DiscussionIn this study, we employed a mouse model of CIN to demonstrate that CoCl2 significantly mitigates kidney damage, renal tubular damage, and epithelial cell death associated with CIN. Subsequently, we conducted quantitative proteomics and bioinformatics analyses to identify and examine key proteins linked to contrast-induced renal damage. Western blotting assays validated changes in candidate protein expression identified through proteomics. Utilizing erastin to induce ferroptosis, we elucidated the interplay between Hp and ferroptosis. Lastly, we employed the HK-2 CIN cell model and adenovirus-overexpressing Hp to confirm that CoCl2 safeguards renal epithelial cells from contrast-induced injury by attenuating ferroptosis, in which Hp likely participates by influencing intracellular ferrous concentration. These findings offer theoretical backing for CoCl2's protective effect on renal tubules after CIN.
The pathogenesis of CIN is intricate and encompasses hemodynamic shifts, ischemia, oxidative stress, and inflammation. Administration of CM prompts the synthesis of vasoconstrictors (e.g., endothelin, adenosine, thromboxane A2) and a reduction in vasodilators (e.g., nitric oxide), leading to renal blood vessel contraction, potentially resulting in ischemic necrosis of renal tubular cells.21 High-viscosity plasma induced by hypotonic contrast agents can lead to renal tubule damage through hypoperfusion and tubule blockage.22 Contrast agents directly inflict toxic damage on renal tubular cells, causing calcium ion accumulation, ROS-mediated DNA fragmentation, mitochondrial dysfunction, and various forms of cell death.8,23 Mitochondrial damage stimulates ROS production, causing oxidative DNA damage and escalating the production of inflammasomes containing protein three pyrimidine domain (NLRP3).24 Contrast agents also induce renal hypoxia, redox reaction imbalances, and increased ROS levels.25,26 Recent studies have highlighted CM's capacity to induce ferroptosis in renal tubular epithelial cells and kidney tissues, characterized by lipid metabolite homeostasis disruption and reduced GSH and GPX4 levels.27,28 Given these findings and the perturbations in redox reactions, ROS, and oxidase, we focused on ferroptosis, which is deemed crucial in CIN.29
Renal ischemia and hypoxia ensuing from CM administration underlie CIN. Hypoxic preconditioning enhances cellular tolerance to hypoxic stress, mitigating the adverse effects of hypoxia on cells.11 CoCl2, a HIF-1α activator, safeguards against renal tubular damage.12 A study demonstrated that CoCl2 treatment of HK-2 cells curtailed inflammation and ROS production.13 ROS significantly impact CIN, with their accumulation pivotal for cellular ferroptosis. This prompts a pertinent query: does CoCl2's hypoxic effect shield renal tubular cells and impact ferroptosis? We addressed this issue by conducting a TMT proteomic analysis of kidney tissue from CIN-afflicted mice treated with CoCl2. Subsequently, traditional molecular experiments identified Hp as a potential key DEP. Western blotting and qPCR findings correlated with TMT-based proteomics outcomes. While Hp was prominently expressed in the CIN model, CoCl2 treatment attenuated its expression.
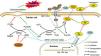
Our in vivo and in vitro findings seemingly juxtaposed each other. Drawing from previous research and our observations, we propose the following rationale (summarized in Fig. 7): in vivo, under physiological conditions, hemoglobin and heme release Fe2+ into cells, fostering the Fenton reaction and elevating •OH and PLOOH (phospholipid hydroperoxides) levels.30 GPX4 and GSH counterbalance this process, reducing PLOOH to PLOH (phospholipid alcohols), thereby sustaining equilibrium.31 Contrast agents diminish GPX4 and GSH levels, instigating ferroptosis in renal tubular cells.32 During ferroptosis, Hp strongly binds to free Hb, curbing its oxidative activity, its role in ferroptosis, and the resulting renal injury.33,34 Hb could also suppress iron accumulation in tubular cells, thus, ameliorating Fenton reaction-related iron storage abnormalities, ROS production, and/or ultimate ferroptosis.35,36 After CoCl2 treatment, reduced iron-transferrin binding results in decreased intracellular iron levels,37 consequently mitigating intracellular Fe2+-induced ferroptosis. This likely causes a decline in required Hp, potentially due to a direct reduction in hepatocyte-derived Hp (given the liver's primary Hp source,38 forming a systemic feedback regulation mechanism). Supporting this speculation, reports indicate decreased plasma haptoglobin levels post-CoCl2 treatment.32 Conversely, Co (II) can mimic hypoxia-like phenotypes by stabilizing HIF-1α,37 possibly leading to enhanced Hp transcription and expression in cobalt-induced tubular cells.39In vitro, tubular cells directly acquire intracellular iron from the culture medium (typically containing Fe3+ and Fe2+), sans Hb, resulting in a disparity from the in vivo environment. This highlights unknown pathways governing Hp-mediated ferroptosis regulation in tubules post-CoCl2 treatment. Notably, altered levels of other ferroptosis-related proteins were observed (Fig. 4), warranting further investigation.

Potential mechanism of Hp's involvement in CIN treatment using CoCl2. Co: cobalt; DMT1: divalent metal transporter 1; GPX4: glutathione peroxidase 4; GSSG: glutathione disulfide; Hb: hemoglobin; Hp: haptoglobin; HO: heme oxygenase; PLOH: phospholipid alcohols; PLOOH: phospholipid hydroperoxides; TF: transferrin; TfR1: transferrin receptor 1; ZIP: zinc-regulated transporter/iron-regulated transporter-like protein. Solid line: in vivo; dotted line: in vitro.
Due to ferroptosis's complexity and resource limitations, our study had some shortcomings. First, other crucial molecules (Cyp2d22, Pla2 g7, Sqle, Cyp4a14), important in ferroptosis, were not explored. Second, the small animal sample size may have introduced data heterogeneity and invalidated conclusions. Additionally, we omitted the group of mice solely treated with CoCl2 in the absence of iohexol. This omission could complicate the understanding of reciprocal iohexol and CoCl2 effects, necessitating a cautious interpretation.
Considering multiple molecular studies on CoCl2's CIN treatment potential, and despite these limitations, CoCl2 might have promising clinical prospects. However, an essential consideration emerges: CoCl2's carcinogenicity arises from its capacity to elevate hypoxia-inducible factor-1α (HIF-1α) expression, a pivotal regulator of tumor survival, metastasis, and angiogenesis.40,41 Furthermore, CoCl2 and exogenous genotoxin co-exposure could enhance genetic instability.42 Consequently, the clinical use of CoCl2 for CIN treatment requires refinement.
ConclusionIn summary, our study revealed that CoCl2 reduced markers of kidney damage post-CIN and alleviated renal tubular injury induced by CIN. Quantitative proteomic analysis of kidney tissue from CIN-infected mice treated with CoCl2 unveiled its potential to exert therapeutic benefits via diverse proteins, targets, and pathways within the CIN mouse model. CoCl2's protective role against CIN-induced renal tubular cell death was validated, and these outcomes were linked to Hp-mediated suppression of ferroptosis.
FundingThis research was supported by National Natural Science Foundation of China (Grant Number: 82170713), Natural Science Foundation of Shanghai (Grant Number: 19ZR1433300), Shanghai Municipal Health Commission Research Project (Grant Number: 202040293), and the Project of Medical Guidance (Chinese and Western Medicine) of Science and Technology Commission of Shanghai Municipality (Grant Number: 134119a5700).
Conflicts of interestNone declared.
The language of this study was professionally edited by ExEditing.com.






