




El síndrome de anticoagulante lúpico-hipoprotrombinemia (LAHS, por sus siglas en inglés) es un trastorno caracterizado por el déficit adquirido del factor ii de la coagulación (protrombina) junto con la presencia de anticoagulante lúpico. Se trata de un síndrome extremadamente raro (menos de 100 casos descritos en la literatura)1 en el cual predomina la diátesis hemorrágica, al contrario que en el síndrome antifosfolípido (SAF) caracterizado por un aumento del riesgo trombótico.
El primer caso fue descrito por Rapaport et al.2, en 1960, pero no fue hasta más de 20 años después cuando el estudio de Bajaj et al.3, demostró la presencia de anticuerpos antiprotrombina que, aunque no impiden su activación, sí producen hipoprotrombinemia secundaria al rápido aclaramiento de los complejos antígeno-anticuerpo de la circulación.
El tratamiento más habitual del LAHS consiste en la corticoterapia asociada a otros inmunosupresores (ciclofosfamida, azatioprina o rituximab) con el objetivo de reducir el riesgo de sangrado y eliminar el inhibidor4.
Presentamos a continuación el caso clínico de un paciente varón de 37 años de edad, de nacionalidad búlgara, con historia de nefropatía lúpica diagnosticada en su país de origen en el año 2004. A pesar de recibir tratamiento presentó un curso clínico desfavorable, precisando tratamiento renal sustitutivo 7 años después. Dos años después acude a nuestro centro para continuar con hemodiálisis. Fue valorado por los servicios de reumatología y hematología por trombocitopenia crónica, por la cual recibió tratamiento inmunosupresor con esteroides, inmunoglobulinas y rituximab, además de agonistas del receptor de trombopoyetina (eltrombopag) con una pobre respuesta. Por otro lado, el paciente padecía un SAF con positividad para los anticuerpos anticardiolipina y anti-ß2-microglobulina, que se presentó con varios eventos trombóticos (trombosis de varios accesos vasculares) y un accidente cerebrovascular en año 2014, por lo que se había iniciado tratamiento anticoagulante.
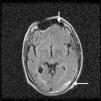
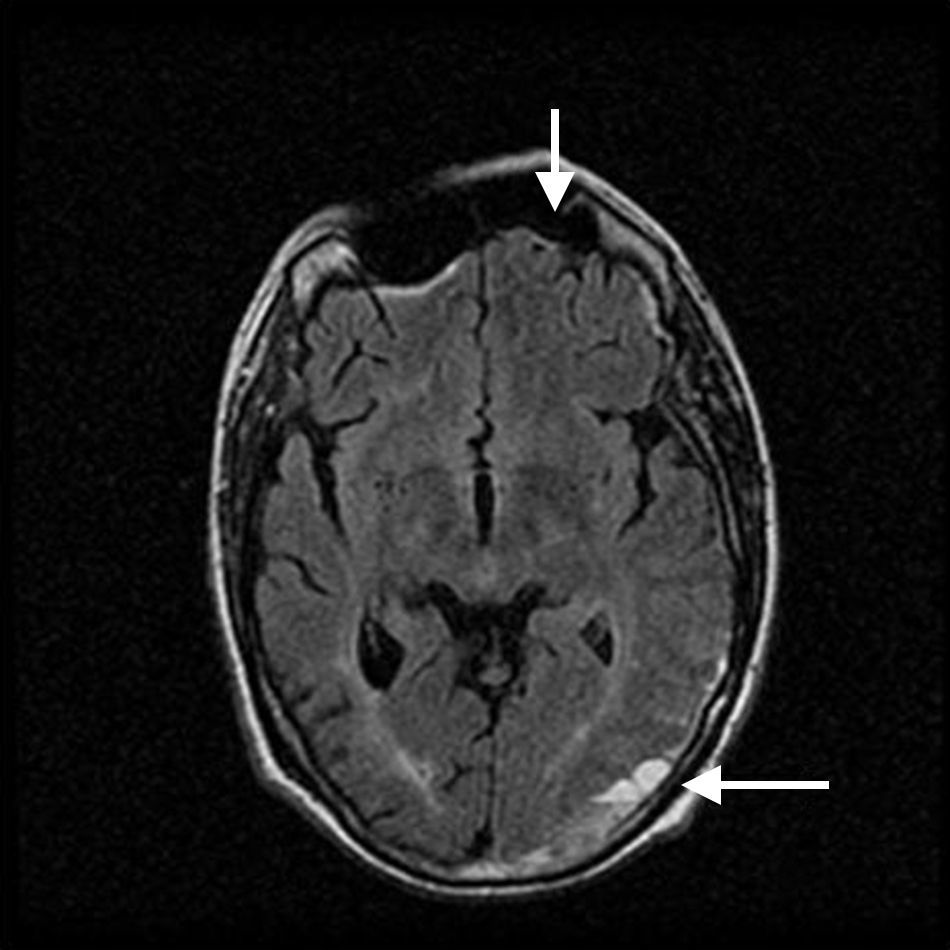
El paciente ingresó en nuestro servicio por un episodio de malestar general y febrícula en el posible contexto de una bacteriemia por catéter central. Se inició tratamiento antibiótico de amplio espectro con una clara mejoría del cuadro y se decidió suspender la anticoagulación temporalmente para proceder al recambio del catéter. Coincidiendo con este proceso presentó un accidente isquémico transitorio caracterizado por parestesias y disminución de fuerza en hemicuerpo derecho junto a afasia motora, por lo que se decidió reiniciar anticoagulación con heparina sódica. La angio-resonancia magnética craneal sorprendentemente mostró, junto a las lesiones isquémicas antiguas, un importante hematoma subdural en la convexidad izquierda con efecto de masa en la región parieto-occipital (fig. 1). Conjuntamente con los servicios de neurocirugía y neurología se decidió suspender la anticoagulación y esperar su evolución clínica y radiológica. Ante la presencia de eventos trombóticos y hemorrágicos se solicitó un nuevo estudio de coagulación que demostró un anticoagulante lúpico, niveles elevados de anticuerpos anticardiolipina y anti-ß2-glicoproteína, y un déficit en la actividad del factor ii de la coagulación (protrombina) que corregía con la mezcla, confirmando la presencia del síndrome de anticoagulante lúpico-hipoprotrombinemia (tabla 1).
Actividad de los factores de coagulación con y sin corrección para el anticoagulante lúpico
| Factores | Actividad factor (%) | Actividad factor corregida (SynthAFax®) (%) | Actividad factor mezcla (%) |
|---|---|---|---|
| Factor II | 25 | – | 70 |
| Factor V | 102 | – | – |
| Factor VII | 136 | – | – |
| Factor X | 114 | – | – |
| Factor VIII | 16 | 83 | – |
| Factor IX | 6 | 96 | – |
| Factor XI | 8 | 94 | – |
| Factor XII | 7 | 98 | – |
Iniciamos tratamiento combinado de anticoagulación e inmunosupresión con bajas dosis de esteroides, ácido micofenólico y rituximab cada 6 meses.
La asociación de anticoagulante lúpico con hipoprotrombinemia es un síndrome raro, pocas veces descrito. Se trata de un síndrome que aparece más frecuentemente en niños y adultos jóvenes, y se asocia comúnmente a infecciones virales y enfermedades autoinmunes (sobre todo al lupus eritematoso sistémico), aunque también se han descrito casos asociados a fármacos o enfermedades tumorales1. El LAHS suele ser autolimitado en el caso de los aparecidos en el contexto de infecciones virales, mientras que en los casos asociados a enfermedades autoinmunes las recidivas son frecuentes a pesar del tratamiento.
Aunque clínicamente se caracteriza por la diátesis hemorrágica (epistaxis y equimosis, las más frecuentes, aunque se han descrito casos de hematuria, hemorragias digestivas e intracraneales entre otras)5. Es cierto que se han descrito varios casos de LAHS asociado a trombosis, algunas múltiples pero, sobre todo en contexto con el inicio del tratamiento frente al inhibidor de la protrombina.
El tratamiento del LAHS se basa en inmunosupresión para evitar los eventos hemorrágicos e intentar eliminar el inhibidor del factor ii4. Se ha descrito un aumento de eventos trombóticos en pacientes con LAHS, al tiempo que la inmunosupresión hace disminuir los niveles de inhibidor, pero no de anticoagulante lúpico5. No existen guías que indiquen cuál es el mejor tratamiento para el LAHS, basándose la mayoría en un tratamiento con corticoides más algún otro inmunosupresor.
En conclusión, en pacientes con nefropatía lúpica con la combinación de procesos trombóticos y hemorrágicos debemos sospechar de la presencia de este síndrome. Es fundamental su diagnóstico y un tratamiento basado en inmunosupresión de cara a controlar sus manifestaciones clínicas.
Conflicto de interesesLos autores declaran que no tienen conflictos de intereses potenciales relacionados con los contenidos de este artículo.



