



Vascular calcification is a pathology characterized by the deposition of calcium-phosphate in cardiovascular structures, mainly in the form of hydroxyapatite crystals, resulting in ectopic calcification. It is correlated with increased risk of cardiovascular disease and myocardial infarction in diabetic patients and in those with chronic kidney disease (CKD). Vascular smooth muscle cells are sensitive to changes in inorganic phosphate (Pi) levels. They are able to adapt and modify some of their functions and promote changes which trigger calcification. Pi is regulated by parathyroid hormone and 1,25-dihydroxyvitamin D. Changes in the transport of Pi are the primary factor responsible for the regulation of Pi homeostasis and the calcification process.
Synthesis of calcification inhibitors is the main mechanism by which cells are able to prevent vascular calcification. Extracellular pyrophosphate (PPi) is a potent endogenous inhibitor of calcium-phosphate deposition both in vivo and in vitro. Patients with CKD show lower levels of PPi and increased activity of the enzyme alkaline phosphatase. Numerous enzymes implicated in the metabolism of PPi have been associated with vascular calcifications. PPi is synthesized from extracellular ATP by nucleotide pyrophosphatase/phosphodiesterase from extracellular ATP hydrolysis. PPi is hydrolyzed into Pi by tissue-nonspecific alkaline phosphatase. ATP can be hydrolyzed to Pi via the ectonucleoside triphosphate diphosphohydrolase family. All these enzymes must be in balance, thereby preventing calcifications. However, diseases like CKD or diabetes induce alterations in their levels. Administration of PPi could open up new treatment options for these patients.
La calcificación vascular es una enfermedad caracterizada por depósitos de fosfato de calcio, principalmente hidroxiapatita, en las estructuras cardiovasculares, dando como resultado calcificaciones ectópicas. Se ha relacionado con un mayor riesgo de padecer enfermedades cardiovasculares e infarto de miocardio en pacientes diabéticos y enfermos renales crónicos. Las células del musculo liso vasculares son sensibles a cambios en los niveles de fosforo inorgánico (Pi), adaptando y modificando su función y promoviendo cambios que desencadenan calcificaciones. El Pi es regulado por hormona paratiroidea y vitamina D. Cambios en el transporte de Pi son el primer factor responsable en la regulación de la homeostasis del Pi y el proceso de calcificación.
La síntesis de inhibidores de calcificación es el principal mecanismo por el que las células nos protegen frente a la calcificación vascular. El pirofosfato extracelular (PPi) es un potente inhibidor endógeno de los depósitos de fosfato-calcio, tanto in vivo como in vitro. Enfermos renales crónicos muestran bajos niveles de pirofosfato y una mayor actividad de la enzima fosfatasa alcalina. Diversas enzimas relacionadas con el metabolismo del PPi extracelular se han relacionado con calcificación vascular. El PPi se sintetiza a partir de ATP extracelular por la nucleótido pirofosfatasa/fosfodiesterasa a partir de la hidrólisis del ATP extracelular. El PPi es hidrolizado a Pi por la fosfatasa alcalina no específica de tejido. El ATP puede ser hidrolizado a Pi por la familia ectonucleósido trifosfato difosfohidrolasa. Todas estas enzimas deben estar en equilibrio, evitando así las calcificaciones; sin embargo, dolencias como la enfermedad renal crónica o la diabetes provocan alteraciones en sus niveles. La administración de pirofosfato puede abrir nuevas vías de tratamiento en estos pacientes.
Vascular calcification (VC)1–3 is a pathology characterized by the deposition of calcium-phosphate in cardiovascular structures, mainly in the form of hydroxyapatite crystals, resulting in vessel thickening and ectopic calcification. Vascular calcification originates in the aortic walls, where calcification takes place in the medial layer of the vessel (Monckeberg's medial sclerosis).4 It is correlated with increased risk of cardiovascular disease and myocardial infarction5 and is associated with aging and diseases like atherosclerosis, hypertension, and metabolic disorders, commonly appearing in diabetic patients and in those with chronic kidney disease (CKD).6 Patients with CKD suffer several metabolic and enzymatic disturbances, which cause an increase in the level of serum inorganic phosphate (Pi) and glucose. High levels of phosphate have been associated with the expression of osteogenic genes, hydroxyapatite formation,7 and overall appearance of vascular calcifications.
Vascular smooth muscle cells (VSMCs) are sensitive to Pi levels, being able to adapt and modify some of their functions. These changes in response to variation in Pi lead to processes that promote calcification. Some of these actions include extracellular matrix calcification, induction of apoptosis, vesicle release, and the expression of osteogenic and chondrogenic genes.5 Many studies have indicated Pi as a major risk factor for cardiovascular diseases in CKD due to the following: 1) Pi is capable of upregulating the expression of osteogenic and chondrogenic genes like Runx2, osteopontin, and alkaline phosphatase (ALP), while downregulating genes that promote smooth muscle cell lineage such as SM22α8,9; 2) Pi may promote the activation of the proapoptotic pathway by reducing gene 6 and its receptor, which is implicated in the antiapoptotic pathway10; and 3) the matrix metalloproteinases MMP-2, MMP-9, and elastin-degrading enzymes can be upregulated by high levels of phosphate.11,12
In addition to the main role of Pi, the importance of calcium-phosphate deposition (CPD) in the vessels in the calcification process has been widely described. High concentration of calcium (Ca2+) interacts with the existing Pi forms (H2PO4−, HPO42−, PO43−) and results in hydroxyapatite crystals (Ca10(PO4)6(OH)), the main form of calcium-phosphate crystals in the vessels.4 Ca2+ is even more relevant given that Ca2+ and Pi are able to interact and regulate each other.13,14 Although calcium deposition increases at the same rate as phosphate. Recent studies have shown that high plasma calcium levels are able to induce CPD even with normal phosphate concentration,15 thereby changing the general scene of VC and opening new pathways for research.
Phosphate homeostasisPhosphate not only plays a role in the calcification process, but it is also involved in physiopathological conditions such as muscle weakness, impaired leukocyte function, and bone mineralization, among others.5 Besides its pathological role, phosphate is essential for maintaining many cellular processes like energy metabolism, cellular signaling, membrane integrity, etc. Widely distributed throughout the body, it is present in plasma in the form of inorganic phosphate, lipid phosphate, and phosphoric ester phosphates.16
Given its critical role in cellular biology and its wide distribution, phosphate needs to be strongly regulated. Under normal conditions, Pi levels are kept stable by the actions of two main hormones: the parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (VitD),17,18 but also fibroblast growth factor 23 (FGF-23) and the enzyme Klotho.19,20 These two main hormones (PTH and VitD) have opposite effects: PTH decreases Pi reabsorption in the kidney while VitD elevates this reabsorption and improves absorption in the intestine.21 Under normal conditions, the amount of phosphate absorbed in the intestine must be equal to the phosphate excreted by the kidney in order to maintain a neutral phosphate status. In addition, the permanent phosphate pool needed for the formation and recycling of the bones must be continually renewed.22 In addition, soluble Klotho protects against Uremic Cardiomyopathy independently of FGF-23 and Pi.23
Changes in the efficiency of Pi transport are the primary factor responsible for the regulation of Pi homeostasis.16 PTH,17 FGF-23,24 and klotho25 promote a decrease in phosphate levels by inactivating and internalizing the specific sodium-phosphate transporter (NaPi cotransporter) in the kidney,26 leading to an increase in the excretion of P and lowering serum levels of Pi. VitD, also known as calcitriol, enhances P absorption in the intestine, raising Pi serum levels.27 Due to renal failure, P absorption and excretion are impaired in CKD patients, leading to high levels of Pi. To maintain Pi within the normal range, FGF-23 and PTH are upregulated, though as the disease progress these systems are unable to uphold correct homeostasis, which results in hyperphosphatemia.24,28,29 These hormonal changes take place in the long run, although in addition to these adjustments there is a mechanism in charge of responding to rapid changes in Pi levels. The intestine contains cells that are able to sense and react to rapid changes in Pi concentration due to diet; however, this system is not able to compensate for the imbalance of Pi in pathologies like CKD.16
All of these hormones that are able to regulate and control Pi levels are called phosphatonins,21 and one particular group of these hormones is becoming more relevant in the context of phosphate homeostasis. The capacity of this sub-group of hormones was discovered in 1994 in patients with tumor-induced osteomalacia (TIO), a disease characterized by high P wasting.29 Phosphatonins promote phosphaturia by inhibiting and modifying the activity of the sodium-dependent phosphate transporter in response to changes in serum phosphate levels30 in the proximal tubules of the kidney and reducing reabsorptions.31 Factors such as FGF-23, secreted frizzled-related protein 4 (sFRP-4), fibroblast growth factor (FGF-7), and matrix extracellular phosphoglycoprotein (MEPE) are included in this group and have been implicated in the pathology of diseases involving low levels of Pi such as TIO, X-Liked hypophosphatemia (XLH), and autosomal dominant hypophosphatemic rickets (ADHR).32–34 Some of these molecules, like FGF-23 and sFRP-4, are able to modify the activity and synthesis of the main hormone regulator VitD. Together with Pi itself, these hormones can regulate their own expression and activity, creating a complex coordinate system responsible for reacting to variations in Pi levels.
Phosphate transportThe main phosphate transporter family is the sodium-dependent phosphate cotransporter, also known as NaPi. These transporters are glycosylated proteins with 10–12 transmembrane regions, whose function is the input of phosphate into the cell. To date, three families of these transporters have been described. Type I and II are widely expressed between species and are distributed mainly in the kidney and intestinal epithelium, while type III transporters are found in many tissues such as kidney, brain, or liver.35 Type I or SLC17 phosphate transporters are mainly implicated in the transport of organic anions, and although they are not involved in Pi transport, they act in diverse processes like vesicular storage or metabolism.36
NaPi-II or SLC34 cotransporters are formed by 3 isoforms: NaPi-IIa, NaPi-IIb, and NaPi-IIc.37 These play a main role in phosphate homeostasis. NaPi-IIa and NaPi-IIc are located at the apical sites of kidney epithelial cells, where they control the resorption of phosphate.26 NaPi-IIb is found in the intestine and is involved in Pi absorption. Regulation of phosphate levels occurs through differential expression and internalization of these transporters on the tissue.37
The most recently discovered phosphate transporter, the NaPi type III or SLC20 family, is predominantly expressed in VSMC, kidney, brain, heart, liver, lung, and osteoblasts. They were discovered as retroviral receptors38 and were first called Glvr-1 and Ram-1. To date, two subtypes have been described in this family, known as Pit-1 and Pit-239; these may play a major role in differentiation to osteogenic/chondrogenic phenotype and in the mineralization process.40 It has been speculated that NaPi type III plays a major role in the calcification process that takes place in VSMCs.
In the renal epithelium both NaPi-II (NaPi-IIa and NaPi-IIc) and NaPi-III (Pit1 and Pit2) with different expression are found.41 NaPi-IIa represents 95% of all phosphate transport, while NaPi-IIc, Pit1, and Pit2 only represent 5%.26 Even though NaPi-IIa is the primary cotransporter behind Pi transport, other transporters such as Pit2, carry out additional functions in Pi transport in other contexts. Pit2 has demonstrated a role when Pi levels are low and metabolic acidosis conditions are found.26 This specificity is due to the affinity that each transporter has with its substrate. As seen before, phosphate can exist in four different forms depending on pH: H3PO4, H2PO4−, HPO42−, and PO43−. Depending on each transporter's substrate, they will have a different pH specificity.4 This explains the diversity of transporters in kidney and their different expression.
Role of phosphate transport in vascular smooth muscle cell calcificationPrevious work from the group of Giachelli42 associated the inhibition of Pit-1 by phosphonoformic acid (PFA) with cell differentiation, upregulation of osteochondrogenic genes like core-binding factor subunit alpha-1 (Cbfa-1) and osteopontin (OPN), and calcification in VSMC. Subsequent studies have shown43 that PFA is not an effective inhibitor of sodium-phosphate co-transporter type III (Pit1 and Pit2). Pit1 and Pit2 are, however, the main Pi transporters in VSMC,40,42,44 and cannot explain by themselves the increase in Pi levels in VC. Furthermore, Pi transport by Pit-1 and Pit-2 is saturated under physiological concentrations of phosphate.44,15
Besides these data, PFA has been demonstrated to play an important role in preventing vascular calcification, though it is unclear whether it is mediated by Pi transporters. Thanks to the homology between PFA and pyrophosphate (PPi), PFA and different agents with similar structure like bisphosphonates are able to mimic the actions of PPi, inhibiting calcium-phosphate deposition.43,45,46 In addition, several studies have shown that calcium-phosphate deposition is a passive process that no required any cellular activity.15,43 Since calcification process is a passive process, synthesis of calcification inhibitors are the main mechanism by which the cell is able to prevent vascular calcification.47 In extracellular fluids, there are a range of endogenous low and high molecular weight inhibitors, including 1) low molecular weight (such as pyrophosphate and citrate) and 2) small and medium-sized proteins (such as, Matrix Gla Proteins, Fetuin A, and osteopontin).48 PPi has been identified as an potent endogenous inhibitor of Ca2+ deposition both in vivo49,50 and in vitro.43,51,52
PPi metabolismPPi is synthesized by nucleotide pyrophosphatase/phosphodiesterase (NPP) from extracellular ATP, turning this enzyme into the main source of PPi.51,53 Two enzymes of this family have been implicated in PPi metabolism (Fig. 1): NPP1 and NPP3.47 While the only action of NPP1 is the production of PPi by hydrolysis of ATP, NPP3 also can hydrolyze this PPi into Pi, promoting VC.47,54 ATP can follow different routes and be hydrolyzed to Pi via ectonucleoside triphosphatediphosphohydrolase1-3 (eNTPD1-3), diminishing PPi disponibility.4 Products of this metabolism such as ADP or AMP can be hydrolyzed into adenosine by 5′-ectonucleotidase (5NT, also known as CD73).55 Adenosine returns to the cell thanks to equilibrative nucleoside transporter 1 (ENT1)56 and can be used for the renewal of ATP.52
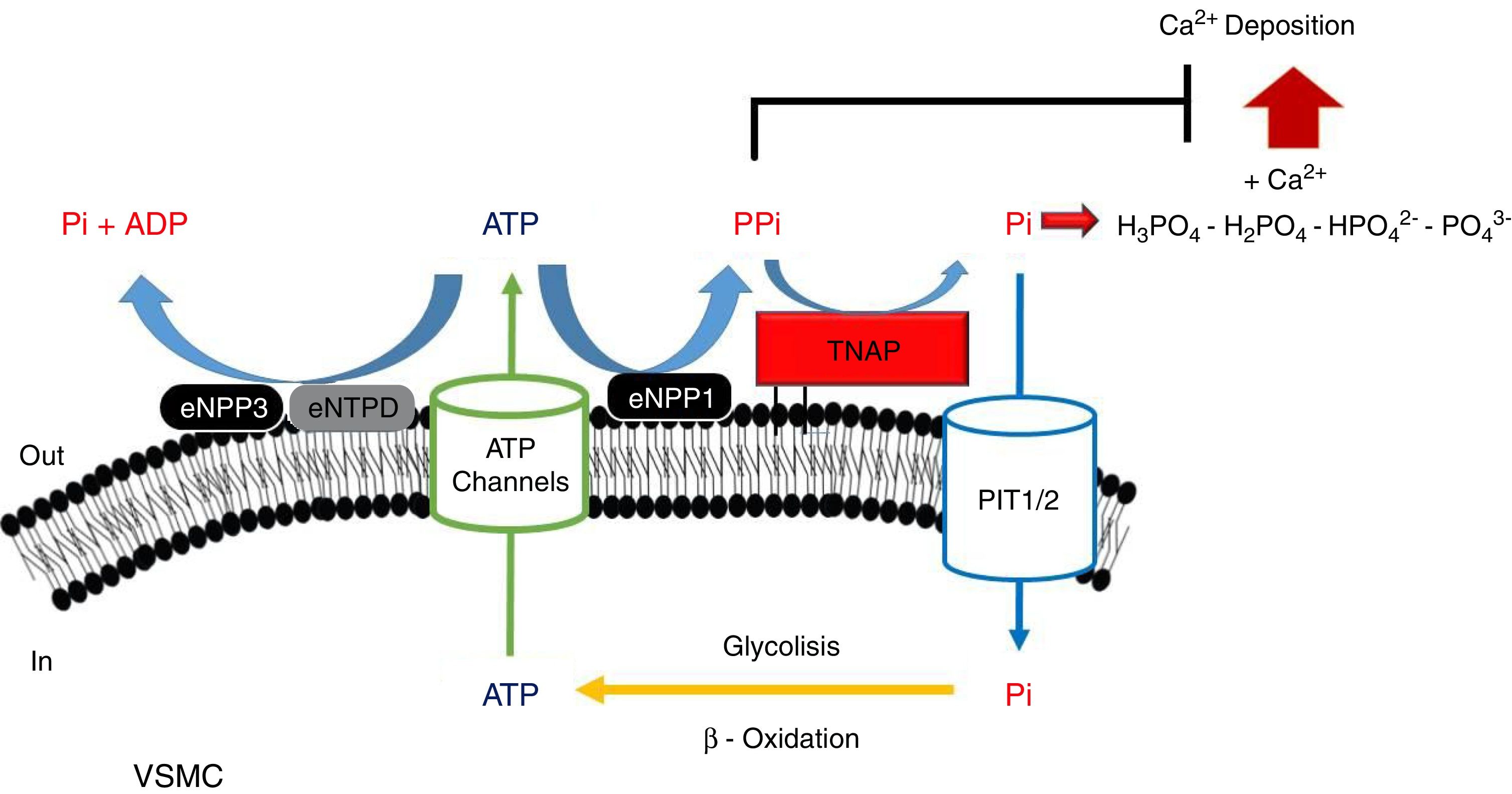
Schematic representation of the ectoenzymes and transporters involved in the extracellular ATP metabolism. Extracellular pyrophosphate (PPi) is the major endogenous inhibitor of ectopic calcification, that is degraded by tissue non-specific alkaline phosphatase (TNAP) and produced during extracellular hydrolysis of adenosine-5′-triphosphate (ATP), via ectonucleotide pyrophosphatase phosphodiesterase 1 (eNPP1). A direct competitor of the substrate for eNPP1 is the ectonucleoside triphosphate diphosphohydrolase (eNTPD) that hydrolyze ATP or ADP releasing inorganic phosphate (Pi) and adenosine-5′-diphosphate (ADP) or adenosine-5′-monophosphate (AMP) respectively. To generate ATP by mitochondria or another metabolic pathway, Pi are recovered from the extracellular space by the sodium-phosphate transporter type III (Pit1/2). To close the cycle of extracellular ATP metabolism, ATP is released by cells via exocytotic mechanisms and via multiple types of membrane channels, including connexin hemichannels, pannexin, cystic fibrosis transmembrane conductance regulator and the sulfonylurea receptor.
Tissue-nonspecific alkaline phosphatase (TNAP), which hydrolyzes PPi into Pi,57 has been one of the most widely studied among all enzymes implicated in VC.5 The role of TNAP in the PPi hydrolysis is the major contributor to this diminishing of its amount.47,49 Studies in CKD patients have shown an increment in its activity due to the clearance of phosphate and hydrogen ions during dialysis session.58
Another mechanism that increases the levels of extracellular PPi is the release of the intracellular component through the putative transporter “progressive ankylosis” (ANK).59 Although this transport outside the cell was never proved, ANK could regulate the extracellular PPi homeostasis, although the exact mechanism is not yet fully understood.4
Role of ATPStudies in experimental models of Hutchinson–Gilford progeria syndrome, a disorder characterized by premature aging and vascular calcification,60 have suggested the role of pyrophosphate and ATP metabolism as potential treatments of VC.49 This syndrome is caused by a mutation in elastin A eliciting the synthesis of progerin, which disrupts the nuclear membrane and can cause genetic alterations.61,62 Among the effects that are related to this mutation we found over expression of osteogenic genes like BMP-2 and Runx2, elevated levels of TNAP and NTPD1; and an overall disruption in ATP metabolism due to a mitochondrial dysfunction.49 PPi treatment is able to revert calcifications in these mouse models, thus providing further support to the hypothesis of PPi as a potent endogenous inhibitor of calcification.63
Several enzymes implicated in ATP hydrolysis have been kinetically characterized, confirming NPP1 as the main source of PPi and TNAP as the enzymes responsible for PPi hydrolysis.47,51,64 With an IC50value lower than PPi (PPi 8.8μmol/L; ATP 3.2μmol/L), ATP alone can act as an inhibitor of calcium-phosphate deposition, and is also more effective in suppressing calcification.51
Genetics in vascular calcificationA number of genetic studies have been performed to understand the processes underlying arterial calcification. Several candidate genes have been proposed to have a role in the calcification process based on animal models or mutations in human diseases.48 Some of the most important discoveries are based on monogenic deficiencies of certain genes found in patients, and these genes help to elucidate the mechanism through which phosphate acts. Several studies found four fundamental genes in four different pathologies characterized phenotypically by ectopic calcifications: pseudoxanthoma elasticum (PXE) and ATP binding cassette subfamily C member 6 (ABCC6) mutation,65 generalized arterial calcification of infancy (GACI) and ENPP1 mutation,66 Pit2 and idiopathic basal ganglia calcification (IBGC),67 and mutation in NT5, which has been implicated in peripheral calcification.55 Moreover, A deficiency in only one of these mediators is able to cause the disease and the pathogenic ectopic calcification. As seen before, all of these mutations affect extracellular ATP/PPi metabolism (Fig. 1), validating the hypothesis of extracellular PPi as an inhibitor of the calcification process.4 In addition, in humans, mutations in the channel core of ANK cause craneometaphyseal dysplasia,68,69 which is characterized by an increased density of craniofacial bones and abnormal modeling of the metaphyses of the tubular bones. Moreover, mutations in the N- and C-terminus of the ANK protein cause chondrocalcinosis,70,71 which is characterized by the deposition of calcium pyrophosphate dehydrate crystals in the joint.
Vascular calcification in CKDVascular calcification is a common pathology in patients with CKD receiving hemodialysis treatment. Prof. O’Neill72 measured PPi in these patients during the pre- and post-hemodialysis periods. These patients showed lower levels of PPi compared to normal subjects, with levels decreasing after dialysis. Not only was extracellular PPi cleared after dialysis; rather, changes in erythrocyte intracellular PPi also took place. These data suggest PPi metabolism as a main component of the calcification process and point to PPi as the primary cause of the effects seen in these patients.
Another study in hemodialysis patients58 showed how physiological alkaline phosphatase (ALP) activity increases postdialysis, as opposed to maximal ALP, which showed no changes. ALP activity increases in parallel with pH, so the slight alkalosis that takes place after dialysis (PreHD 7.25; Post HD 7.45) contributes to the increase in ALP activity.58 Other molecules implicated in PPi metabolism such as ATP and PPi itself exhibited decreased levels after dialysis. Pi acts as a negative feedback of alkaline phosphatase, so the clearance of phosphate, which takes place during hemodialysis, ultimately leads to an increase in ALP activity. Moreover, uremic toxin increases ALP activity and PPi hydrolysis in aortic rats.57
ConclusionPi concentration levels show an important increase in patients with CKD. This increment is associated, mainly, with increments in PTH and FGF-23 levels and reduction in plasmatic levels of VitD and Klotho. Moreover, during dialysis session the levels of plasmatic extracellular PPi are reduced in association with increments in the alkaline phosphatase activity, the main enzyme responsible for degradation of extracellular PPi.
Although alteration in the phosphate homeostasis plays a key role in the process of calcium-phosphate crystal formation, loss of inhibition enhanced the calcification process. Therefore, the homeostasis of phosphate and pyrophosphate should be studied together.
Key concepts- 1.
In chronic renal disease, hyperphosphataemia plays an important role in the development of vascular calcification.
- 2.
The decrease of extracellular pyrophosphate in chronic renal disease, potentiates vascular calcification.
- 3.
Alterations in the balance between phosphate and pyrophosphate may play an important role in the development of vascular calcification.
No conflicts of interest, financial or otherwise, are declared by the authors(s).
We thank Oliver Shaw for English-language revision. This study was supported by grants given from the Spanish Ministerio de Economia y Competitividad (SAF-2014-60699-JIN), Instituto de Salud Carlos III (“Sara Borrell” CD14/00198 postdoctoral contract) to RVB; and Foundation SENEFRO (Spanish Nephrology Society) and FEDER funds PI14/00386 to JE.



