




La activación de la vía alternativa del complemento interviene en el desarrollo de varias enfermedades renales, como el síndrome hemolítico urémico atípico o la glomerulopatía C3. En esta última enfermedad un elevado porcentaje de los pacientes presentan niveles circulantes de un autoanticuerpo denominado C3NeF, causante de la desregulación del complemento a nivel sistémico. En ciertos casos, la presencia de este anticuerpo se asocia con alteraciones en el tejido adiposo, causando lipodistrofia parcial adquirida (síndrome de Barraquer-Simons), una enfermedad ultra-rara que afecta a la distribución del tejido adiposo subcutáneo y que comienza principalmente durante la infancia. Estos pacientes, además de poder presentar los problemas metabólicos asociados al defecto en el tejido adiposo, presentan hipocomplementemia C3 junto con la presencia de C3NeF y, en un 25% de los casos desarrollan una glomerulopatía C3. Aunque se sabe desde hace tiempo cómo la desregulación del sistema del complemento afecta al riñón, se desconoce de forma precisa cómo lo hace en el tejido adiposo; no obstante, su relación está bastante clara. En este artículo se va a describir la relación del sistema del complemento con la biología del tejido adiposo y su patogenia reflejada a partir de la lipodistrofia parcial adquirida.
The activation of the alternative pathway of the complement is involved in the development of several renal diseases, such as atypical haemolytic uremic syndrome and C3 glomerulopathy. In C3 glomerulopathy, a high percentage of patients have circulating levels of the autoantibody called C3NeF, which causes systemic dysregulation of the complement system. In some cases, the presence of this antibody has been related with abnormalities of adipose tissue, causing acquired partial lipodystrophy (Barraquer-Simons syndrome). Acquired partial lipodystrophy is an extremely rare disorder affecting the distribution of subcutaneous adipose tissue and that mainly onsets during childhood. These patients, in addition to possibly presenting with all the metabolic disorders associated with the adipose tissue defect, present with C3 hypocomplementemia and C3NeF and 25% have developed C3 glomerulopathy. Although it has been known for some time how the dysregulation of the complement system affects the kidneys, it remains unknown how it exactly affects adipose tissue; nevertheless, the relationship is quite clear. In this paper, we describe the connection between the complement system with the biology of the adipose tissue and its pathogenesis reflected from acquired partial lipodystrophy.
El sistema del complemento es un componente fundamental de la inmunidad innata, donde juega un papel crucial en la defensa contra infecciones, la eliminación de células apoptóticas, el procesamiento de inmunocomplejos y la modulación de la inmunidad adaptativa. El complemento es un complejo sistema molecular capaz de disparar señales de alarma ante la presencia de agentes extraños al organismo, de discriminar componentes propios y extraños y, mediante un sistema de etiquetado molecular, de identificar estos últimos para su eliminación por opsonofagocitosis o su destrucción mediante lisis celular directa. La activación del complemento tiene lugar a través de 3 vías de activación distintas: la vía clásica, la vía alternativa y la vía de las lectinas (fig. 1). Estas 3 vías, estrechamente relacionadas filogenéticamente, se diferencian en sus mecanismos de activación y en las etapas iniciales, pero todas ellas confluyen en la formación de complejos enzimáticos multimoleculares responsables de la activación del componente C3, denominados convertasas de C31.
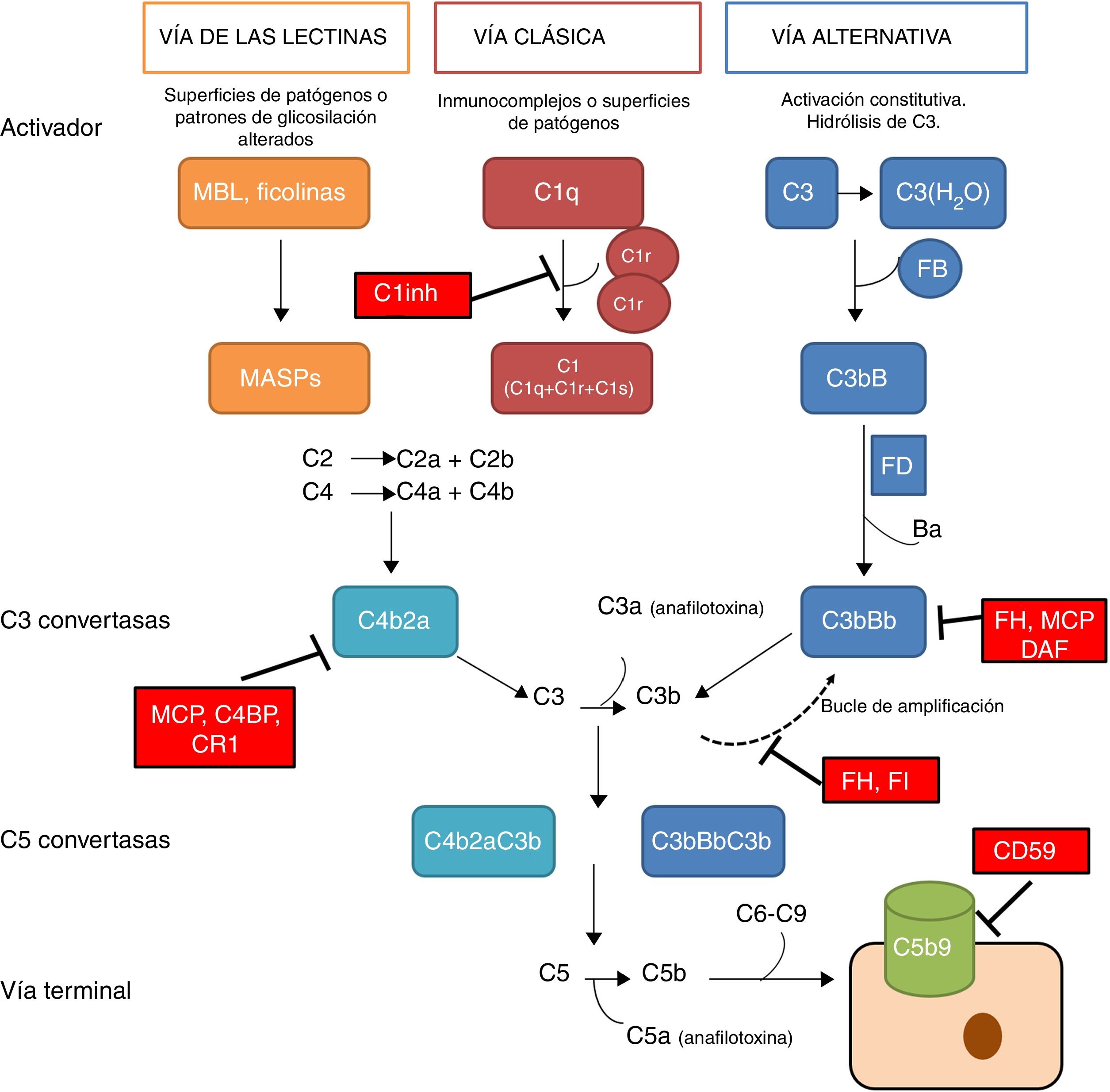
El sistema del complemento. El sistema del complemento puede ser activado por 3 vías. La activación por alguna de ellas lleva a la generación de una convertasa de C3 (C4b2b o C3bBb) que corta C3 en C3a y C3b. El C3b generado por cualquier convertasa puede formar, a su vez, más convertasa de la vía alternativa, a través de la cual se produce la amplificación de la activación del complemento. La unión de una nueva molécula de C3b a las convertasas de C3 les confiere la capacidad de cortar C5 en C5a y C5b. Este último inicia la vía terminal del complemento, que eventualmente lleva a la formación del complejo de ataque a la membrana (C5b-9), lisando las células diana. La activación del complemento está controlada a varios niveles mediante diversas proteínas reguladoras, tanto solubles como de membrana.
La vía clásica se activa principalmente por la unión de C1q a complejos antígeno-anticuerpo y la activación de la vía de las lectinas ocurre fundamentalmente por el reconocimiento de grupos manosa característicos de superficies bacterianas. La activación de estas 2 vías da lugar a la generación de un complejo proteico con actividad enzimática: la convertasa de C3 de la vía clásica/de las lectinas (C4b2b) capaz de activar la molécula de C3, escindiéndola en C3a y C3b.
La vía alternativa es constitutivamente activa, debido a la activación espontánea de C3 en plasma a través del mecanismo «tick-over». La activación de C3 genera los fragmentos C3a y C3b, pudiéndose asociar este último al factor B (FB) para formar la convertasa de C3 de la vía alternativa (C3bB) en estado inactivo. Cada complejo C3bB pasa a su estado activo por medio del corte proteolítico que ocasiona el factor D (FD) sobre FB, originando así los fragmentos Ba y Bb. El complejo resultante, C3bBb, es capaz de amplificar el sistema por medio de un feedback positivo para generar en muy poco tiempo miles de moléculas de C3b1. La convertasa C3bBb que se forma es inestable y requiere estabilizarse por medio del regulador positivo, properdina, el cual incrementa la vida media de este complejo multimolecular y sirve como un foco para la amplificación local del complemento2. Cuando una molécula de C3b se incorpora a una convertasa de C3 se origina la convertasa de C5, capaz de cortar C5 generando C5a, una potente anafilotoxina, y C5b que actúa como iniciador en superficies celulares para conformar junto a C6, C7, C8 y C9 el complejo de ataque a la membrana (C5b9).
La rápida y efectiva disociación del complejo C3bBb y la inactivación de C3b es un paso crítico para la homeostasis del complemento y prevenir el daño sobre superficies o tejidos propios cuando se activa. Estas funciones son llevadas a cabo por una serie de proteínas reguladoras solubles (factor H y factor I) y de membrana (MCP, DAF, CR1 y CD59) (fig. 1). La importancia de la integridad de este sistema se pone de manifiesto porque las deficiencias de alguno de sus componentes pueden causar afecciones importantes que pueden llegar a ser letales. También los problemas surgen por una regulación ineficiente lo que puede ocasionar activación indiscriminada y generación de fragmentos circulantes o lesiones en tejidos3.
Uno de los tejidos que más se ve afectado por una desregulación del sistema del complemento es el riñón. El glomérulo es muy sensible a los efectos inflamatorios como consecuencia de la activación del complemento tanto a nivel sistémico como a nivel local en superficies. Las 2 afecciones que más comúnmente se han visto relacionadas con alteraciones del sistema del complemento son la glomerulopatía C3 (C3G) y el síndrome hemolítico urémico atípico (aHUS). En ambos casos, se han encontrado mutaciones en genes de componentes o reguladores de la vía alternativa, cuyo efecto provoca que la activación de esta vía no pueda ser controlada y el tejido se vea afectado4. Además de las mutaciones, existen autoanticuerpos directamente relacionados con la aparición de estas enfermedades, los cuales tienen un efecto negativo en la correcta regulación de la vía alternativa5. Unos de los autoanticuerpos más conocidos es el factor nefrítico de C3 (C3NeF), un anticuerpo que es capaz de unirse con elevada afinidad a la convertasa de C3 de la vía alternativa (C3bBb) haciendo que sea notablemente más estable y se alargue su vida media (fig. 2). Este anticuerpo es muy común en la C3G, encontrándose en un 80% de los pacientes con enfermedad por depósitos densos y en un 40-50% de los pacientes con glomerulonefritis C36.
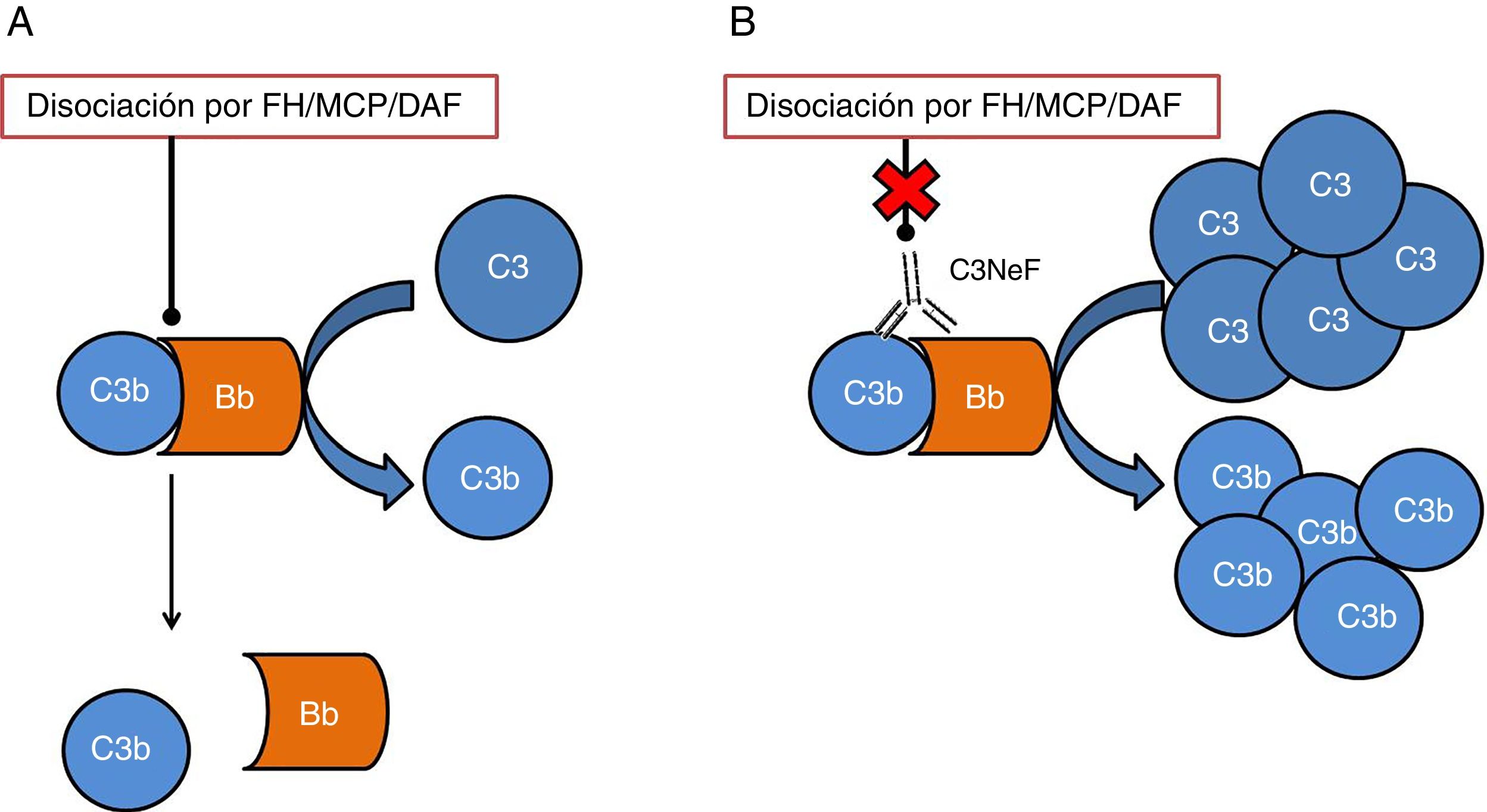
Esquema del efecto de C3NeF sobre la convertasa de C3 de la vía alternativa. A) En condiciones fisiológicas, la convertasa de C3 escinde la molécula de C3 en C3a y C3b, en un proceso regulado por proteínas como factor H (FH), MCP o DAF. B) Cuando C3NeF se une a la convertasa, impide que los reguladores FH, MCP o DAF puedan disociar el complejo, permaneciendo activa durante más tiempo y ocasionando un aumento del consumo de C3.
Otra situación patológica donde C3NeF es altamente prevalente es en la lipodistrofia parcial adquirida (también conocido como síndrome de Barraquer-Simons), un trastorno extremadamente raro del tejido adiposo subcutáneo, donde un 25% de los pacientes desarrollan una C3G a medio-largo plazo7. En este artículo se va a hacer una revisión del papel del sistema del complemento en la biología del tejido adiposo y en la patogenia de la lipodistrofia parcial adquirida, así como su relación con la C3G, y en último lugar se discutirán las pautas terapéuticas existentes, incluida la terapia anticomplemento.
El sistema del complemento en la biología del adipocitoEl grupo del doctor Spiegelman demostró que los adipocitos múridos sintetizaban y secretaban una proteína que llamaron adipsina, la cual tenía unas funciones relevantes para el desarrollo del adipocito8,9. Años después, en humanos se encontró una proteína con una homología con adipsina superior al 98%. Esta proteína resultó ser FD, de la cual ya se conocía que tenía unas funciones clave en la activación de la vía alternativa del complemento10. Además, a diferencia de la mayoría de las proteínas del complemento, que se sintetizan en el hígado, un 95% de FD circulante se sintetiza en el tejido adiposo11. También el adipocito sintetiza C3, FB12 y properdina13. Estos datos manifiestan la importancia de la vía alternativa en el tejido adiposo, siendo los adipocitos capaces de generar una convertasa de C3 (C3bBb) en su exterior, enriqueciendo el espacio extracelular con C3a, una potente anafilotoxina pro-inflamatoria (fig. 3A). En un estudio independiente Sniderman y Cianflone describieron la presencia de un factor en suero capaz de estimular la síntesis de triglicéridos en el tejido adiposo humano, el cual recibió el nombre de Acylation Stimulating Protein (ASP). Después de secuenciarla, descubrieron que correspondía a C3desArg14, un producto de la degradación de C3a por la carboxipeptidasa N. Posteriores estudios revelaron que los adipocitos presentaban el receptor de complemento C5AR2 en la superficie de la membrana plasmática, el cual une ASP para estimular la síntesis de triglicéridos y promover la maduración de los pre-adipocitos en modelos múridos in vitro15,16 (fig. 3B).
![Papel del sistema del complemento en la biología del adipocito. A) Los adipocitos secretan componentes del complemento como C3, factor B (FB) y factor D (FD, adipsina) y son capaces de generar una convertasa de C3 (C3bBb) de la vía alternativa en sus inmediaciones. B) C3a se escinde por activación de la molécula de C3. A su vez C3a se convierte en C3adesArg (acylation stimulating protein [ASP]) por acción de la carboxipeptidasa N del tejido adiposo. C3adesArg actúa como ligando para su receptor, C5AR2, el cual se localiza en la superficie de los adipocitos, y cuya función principal es señalizar para estimular la síntesis de triglicéridos durante la maduración del tejido adiposo.](https://static.elsevier.es/multimedia/02116995/0000003800000003/v2_201810040615/S0211699517302278/v2_201810040615/es/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92lL8PRnKGMnsiVXV6EVJ5QRkjJZIKj2umwUyY+cL6K89r2TdTkn+7Tb6nUMw4PbSRJI77zJ0KSnTZ2/9BoDFGMF2LKeZU/JCM/o8pcPcsOYAPyeP+vtfzKlMDHWS1OFGZfGSOss06RvoFmE6617FBnG4yhMzhWQigiLvD7+CQCZjRIXjXzF69Rps5sYmkRyDpu+WHiwxrt1StSMpeS0q/fiFgjvjcCyBOBBdrpKlwfEZcHPvx/fDxJBmqi9dMORMDqCvQrNPjZOTC7K6yqqf00=)
Papel del sistema del complemento en la biología del adipocito. A) Los adipocitos secretan componentes del complemento como C3, factor B (FB) y factor D (FD, adipsina) y son capaces de generar una convertasa de C3 (C3bBb) de la vía alternativa en sus inmediaciones. B) C3a se escinde por activación de la molécula de C3. A su vez C3a se convierte en C3adesArg (acylation stimulating protein [ASP]) por acción de la carboxipeptidasa N del tejido adiposo. C3adesArg actúa como ligando para su receptor, C5AR2, el cual se localiza en la superficie de los adipocitos, y cuya función principal es señalizar para estimular la síntesis de triglicéridos durante la maduración del tejido adiposo.
Las lipodistrofias son un conjunto heterogéneo de afecciones ultra-raras caracterizadas por el déficit específico de tejido adiposo en ausencia de deprivación nutricional o estado catabólico7. La pérdida de tejido adiposo puede afectar al cuerpo entero (generalizadas), solo a ciertas regiones bien delimitadas (parcial), o a pequeñas áreas bajo la piel (localizadas). Se han reportado más de 1.000 casos en el mundo, con una prevalencia menor de 1:1.000.000, aunque es probable que esté subestimado. Los tipos anteriormente mencionados se pueden agrupar a su vez en función de su etiología como: lipodistrofias genéticas (familiares) o adquiridas17. A diferencia de las lipodistrofias localizadas, las formas parciales o generalizadas predisponen a los pacientes a desarrollar resistencia a la insulina y a las complicaciones asociadas a este estado como diabetes mellitus, hipertrigliceridemia, esteatosis hepática, ovario poliquístico y acantosis nigricans. La severidad de estas complicaciones dependerá del subtipo, la extensión de pérdida de grasa, la edad y el sexo18.
Los principales subtipos de lipodistrofias incluyen la lipodistrofia generalizada congénita, lipodistrofia parcial familiar, lipodistrofia generalizada adquirida y la lipodistrofia parcial adquirida. Existen otras enfermedades sistémicas que se asocian a lipodistrofia, como los síndromes progeroides y las enfermedades autoinflamatorias18.
Aunque el estudio genético no es necesario para el diagnóstico, es muy útil para identificar subtipos de lipodistrofias familiares. Las pruebas genéticas pueden ayudar a identificar a los miembros de la familia en riesgo, especialmente para los subtipos de lipodistrofia asociados con fenotipos físicos sutiles o aquellos con alto riesgo de morbimortalidad (por ejemplo, cardiomiopatía y arritmias). Un resultado negativo no implica que no se padezca la enfermedad, pues hay formas de lipodistrofias cuya base genética es desconocida.
La mayoría de los genes donde se han descrito mutaciones codifican para proteínas implicadas en la diferenciación/supervivencia del adipocito, o en el proceso de formación de la gota lipídica. La lipodistrofia generalizada congénita está causada por mutaciones autosómicas recesivas en AGPAT2, BSCL2, CAV1 y PTRF. Por otra parte, la lipodistrofia parcial familiar está causada principalmente por mutaciones autosómicas dominantes en LMNA, PPARg, PLIN1 y AKT2; aunque existen 2 tipos con mutaciones recesivas en CIDEC y LIPE17. En la tabla 1 se hace referencia a todos los subtipos de lipodistrofias congénitas existentes, el gen asociado y su principal función.
Principales lipodistrofias congénitas: clasificación, genes causales y función principal
| Gen | Función principal | ||
|---|---|---|---|
| Lipodistrofia generalizada congénita | Tipo 1 (OMIM# 608594) | AGPAT2 | Enzima que juega un papel fundamental en la biosíntesis de triglicéridos y fosfolípidos |
| Tipo 2 (OMIM# 269700) | BSCL2 | Está implicada en la fusión de las gotas lipídicas y la diferenciación adipogénica | |
| Tipo 3 (OMIM# 612526) | CAV1 | Componente de las caveolas implicado en la translocación de ácidos grasos y otros lípidos hacia las gotas lipídicas | |
| Tipo 4 (OMIM# 613327) | PTRF | Implicado en la biogénesis de las caveolas, regulando la expresión de las caveolinas 1 y 3 | |
| Lipodistrofia parcial familiar | Tipo 1 (OMIM% 608600) | Desconocido | |
| Tipo 2 (OMIM# 151660) | LMNA | Componente estructural de la envoltura nuclear | |
| Tipo 3 (OMIM# 604367) | PPARG | Regulador principal de la diferenciación del adipocito | |
| Tipo 4 (OMIM# 613877) | PLIN1 | Componente estructural de la gota lipídica, participa en la regulación de la lipólisis | |
| Tipo 5 (OMIM# 615238) | CIDEC | Componente estructural de la gota lipídica, participa en la regulación de la lipólisis | |
| Tipo 6 (OMIM# 615980) | LIPE | Regulación de la lipólisis | |
| Asociado a mutaciones en AKT2 | AKT2 | Implicado en la transmisión intracelular de la señal por acción de la insulina a través de su receptor |
AGPAT2: 1-acylglycerol-3-phosphate O-acyltransferase 2; AKT2: v-akt murine thymoma viral oncogene homolog 2; BSCL2: Berardinelli-Seip congenital lipodystrophy 2; CAV1: caveolin 1; CIDEC: cell death-inducing DFFA-like effector c; LIPE: hormone sensitive lipase; LMNA: lamin A/C; PLIN1: perilipin; PPARG: peroxisome proliferator-activated receptor gamma; PTRF: polymerase I and transcript release factor.
Las lipodistrofias adquiridas son más frecuentes que las de tipo familiar. La lipodistrofia parcial adquirida (OMIM%613913) fue la primera que se describió hace más de 130 años19. Este síndrome se caracteriza por la desaparición del tejido adiposo subcutáneo en la cara, extremidades superiores y tronco, hipertrofiándose la grasa de las extremidades inferiores. Las mujeres se ven afectadas hasta 4 veces más que los varones. La media de edad de inicio de la lipodistrofia se sitúa en los 7 años. En cuanto a la prevalencia de alteraciones metabólicas, existe algo de controversia al respecto, pero lo que está claro es que esta lipodistrofia se asocia con mucha menor frecuencia a estos trastornos en comparación a otras lipodistrofias parciales familiares (43%), y con mucha diferencia a todos los tipos de generalizadas (50-80%)20. En este tipo de lipodistrofia un 75-90% de los pacientes presentan niveles muy bajos del componente C3 del sistema del complemento. A su vez, y muy relacionado con lo anterior, más del 80% de los pacientes presentan títulos detectables de C3NeF18. En este sentido, dado que la presencia de este autoanticuerpo está fuertemente asociada a la aparición de C3G, es lógico que una cuarta parte de los pacientes con esta lipodistrofia acaben desarrollando la glomerulopatía. La relación entre la enfermedad renal y la lipodistrofia parcial se describió por Gellis et al. (1958)21, y posteriormente se asoció con la glomerlonefritis mesangio-capilar por otros autores22. Dentro del espectro de la C3G, la forma de presentación más común es la enfermedad por depósitos densos20, aunque existe algún caso en el que el estudio histológico desveló una nefropatía IgA23. Es interesante, sobre todo para un adecuado control clínico de estos pacientes, tener en cuenta que el intervalo de tiempo desde el inicio de la lipodistrofia hasta el desarrollo de la nefropatía es de aproximadamente 8 años20. En otras series lo elevan hasta los 10 años, e incluso destacan algún caso donde la aparición de la glomerulopatía se retrasó hasta 20 años22. Por tanto, es recomendable vigilarlos para prevenir o actuar con prontitud ante la aparición de la nefropatía.
En las cohortes de C3G solo un bajo porcentaje de los mismos presentaría a su vez lipodistrofia parcial adquirida. Sin embargo entre los pacientes con lipodistrofia, la frecuencia de encontrar a su vez C3G se incrementa notablemente. En un estudio con 125 pacientes con glomerulonefritis membranoproliferativa, 11 tuvieron hipocomplementemia C3, pero solamente 2 fueron diagnosticados de lipodistrofia24. El único nexo entre ambas afecciones es la hipocomplementemia C3 y la presencia de C3NeF. En este sentido se hipotetizó que una desregulación de la vía alternativa como consecuencia de la presencia de C3NeF pudiera ocasionar algún trastorno de la grasa, debido principalmente a una citotoxicidad dependiente del complemento. Esta posible relación fue analizada por Mathieson y su equipo, los cuales comprobaron que un suero de un paciente con lipodistrofia parcial adquirida, positivo para C3NeF, era capaz de lisar los adipocitos, mientras que el de un donante sano era incapaz de hacerlo25. Por tanto, el mecanismo patogénico que actualmente está aceptado es que C3NeF es capaz de aprovechar la capacidad del adipocito para formar convertasas de C3 en sus inmediaciones e inducir una lisis mediada por complemento (fig. 4). A pesar de ello, el mecanismo fisiopatológico del C3NeF en la lipodistrofia parcial adquirida es desconocido y en cierta medida, discutible. Se sabe que C3NeF no es un autoanticuerpo exclusivo de este síndrome, sino que aparece también, y en mayor proporción, en otras situaciones patológicas no asociadas a la lipodistrofia. Además, muchos autores dan por hecho que existe un patrón de síntesis de FD que va de más a menos desde el tejido subcutáneo de la parte superior del cuerpo a la parte inferior, y que C3NeF destruye el adipocito en las zonas donde más FD se sintetiza; sin embargo, esto no se ha demostrado. Por tanto, deben existir otros factores fisiopatológicos que intervienen en el cuadro clínico y que son desconocidos o bien el C3NeF en estos pacientes tiene unas características bioquímicas diferentes a las del resto de pacientes sin lipodistrofia.
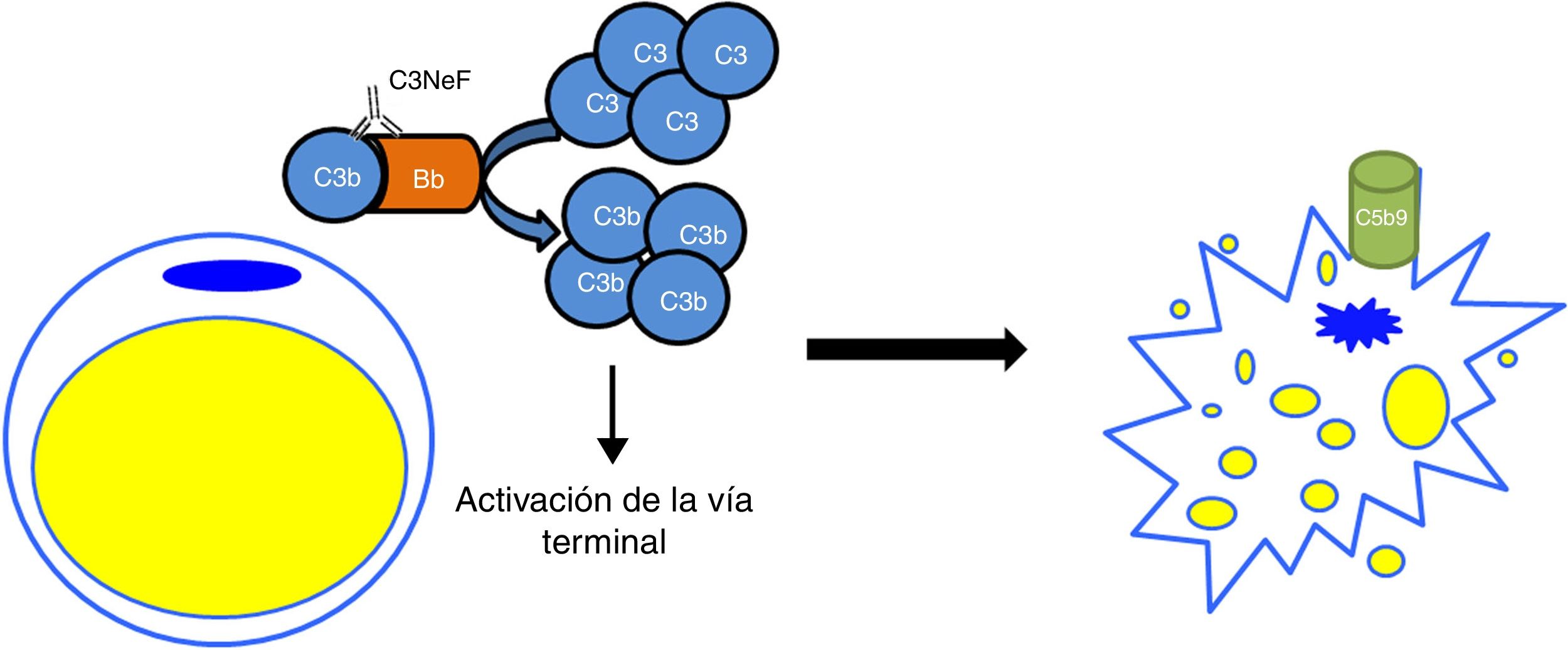
Mecanismo patogénico del C3NeF en la lipodistrofia parcial adquirida. C3NeF se va a servir de las proteínas del complemento que el adipocito sintetiza y va a conducir a una activación descontrolada de la vía alternativa en las inmediaciones de la célula. Esta situación progresa hasta la activación de la vía terminal y la formación del complejo de ataque a la membrana en la superficie del adipocito, provocando su lisis.
La lipodistrofia parcial adquirida no tiene una base genética clara. Los casos descritos son esporádicos, sin un patrón de herencia mendeliano. Salvo excepciones, lo más habitual es encontrar familias con un descendiente afecto y progenitores y otros hermanos sanos. Un caso muy interesante, descrito en España, fue el de una familia en la que 2 hermanas HLA idénticas presentaron lipodistrofia parcial adquirida, infecciones recurrentes, alteraciones del complemento y nefropatía26; sin embargo, no se realizó un análisis de exoma completo para encontrar algún marcador genético común relacionado con la ausencia de tejido adiposo. Otro trabajo elaborado por un grupo británico describió por primera vez una familia con lipodistrofia parcial adquirida y C3G en la que la primera segrega con la presencia de C3NeF y la segunda con una mutación en el gen FH. El análisis de exoma no desveló ninguna variante genética en común a todos los miembros afectados por la lipodistrofia27.
Se ha asociado la susceptibilidad a desarrollar lipodistrofia parcial adquirida con mutaciones en heterocigosis en el gen que codifica la proteína de la envuelta nuclear denominada lamina B2 (LMNB2; OMIM#608709)28. Los resultados en 4 pacientes demostraron que 2 de las 4 variantes encontradas también estaban presentes en donantes sin la enfermedad. Además, el patrón de pérdida de grasa afectaba a las rodillas, los muslos y la región del glúteo, lo cual es muy atípico en esta lipodistrofia, que afecta principalmente a la cabeza, el cuello, el tronco y las extremidades superiores y no lo hace en las extremidades inferiores. Ninguno de los pacientes presentó hipocomplementemia C3, ni se detectó C3NeF. Tres tuvieron diabetes mellitus y todos presentaron hipertrigliceridemia, siendo esto muy poco frecuente en la lipodistrofia parcial adquirida. Finalmente, cabe destacar que no hubo segregación de estas variantes en los miembros de la familia. Por tanto, sin más datos ni experiencia, es altamente improbable que las variantes en el gen LMNB2 puedan asociarse a la lipodistrofia parcial adquirida.
Tratamiento en pacientes con lipodistrofia parcial adquirida: trasplante autólogo de tejido adiposo y terapia anticomplementoRecientemente se ha publicado una guía donde se recogen las pautas para el diagnóstico y el manejo clínico de pacientes con síndromes lipodistróficos infrecuentes29. Este trabajo, donde han participado sociedades de endocrinología de todo el mundo y otros comités de expertos, supone una herramienta de un gran valor a la hora de enfrentarse a pacientes con estas afecciones.
El abordaje terapéutico en esta afección se basa exclusivamente en la reparación cosmética. El trasplante autólogo de tejido adiposo de zonas no afectadas está considerado un tratamiento seguro y eficaz para la restauración del contorno facial y del volumen mamario, pues suelen ser las regiones más afectadas en estos pacientes30. Este tratamiento está justificado ya que como consecuencia de la pérdida del tejido adiposo la mayoría acaban padeciendo un trastorno psicológico grave asociado a la alteración de su aspecto físico. La experiencia al respecto es poca, de modo que es difícil predecir la duración que puede llegar a tener el injerto o cuántas intervenciones son necesarias para conseguir una restauración aceptable; pero los efectos positivos para los pacientes están claramente demostrados por lo que se recomienda encarecidamente valorar este abordaje en todos los casos. Durante la adolescencia aumenta el riesgo de que padezcan trastornos psicológicos que pueden dificultar su vida personal y social una vez sean adultos, por lo que los expertos afirman que debería considerarse la restauración con tejido adiposo autólogo muy pronto, si es posible, durante la infancia29,30.
Los avances en el conocimiento del funcionamiento del sistema del complemento han sido de gran relevancia para la comprensión y el papel de este en el desarrollo de numerosas enfermedades. Su implicación, tanto directa como indirecta en la patogenia de algunas enfermedades, ha abierto una vía para investigar cómo modular la respuesta del sistema del complemento. Actualmente existen algunos fármacos con una eficacia altamente probada en mitigar o restaurar los daños producidos por la desregulación del complemento. Un ejemplo es el uso terapéutico del anticuerpo anti-C5, denominado eculizumab (Soliris®, Alexion Pharmaceuticals), en enfermedades mediadas por el sistema del complemento como hemoglobinuria paroxística nocturna o el síndrome urémico hemolítico atípico31. Existe el caso de una paciente de 14 años con C3G (enfermedad por depósitos densos) asociada a lipodistrofia parcial adquirida que mejoró su función renal tras recibir tratamiento con eculizumab32. En lo que respecta a la C3G, los datos de la eficacia del tratamiento con eculizumab son muy heterogéneos y poco concluyentes, lo que ha impulsado el desarrollo de numerosos ensayos clínicos31. En el caso citado con anterioridad se demuestra que el bloqueo de C5 es de gran utilidad cuando otros tratamientos más generales, como la terapia con corticoides o la plasmaferesis, son insuficientes. Sin embargo, no existen datos en la literatura que demuestren que el tratamiento anticomplemento tenga algún beneficio sobre el tejido adiposo ni las alteraciones metabólicas (si las hubiera) en pacientes con lipodistrofia parcial adquirida.
ConclusionesLas lipodistrofias, tanto las de tipo hereditario como adquirido, son enfermedades extremadamente raras, lo cual lleva implícito que son un verdadero enigma cuando un paciente llega a una consulta médica. En el síndrome de Barraquer-Simons (lipodistrofia parcial adquirida), en el cual se centra la presente revisión, la gran mayoría de los pacientes presentan hipocomplementemia C3 debido a la aparición del C3NeF, un potente desregulador de la vía alternativa. Por su parte, C3NeF se asocia en la mayor parte de los casos con la aparición de C3G.
Actualmente se conoce perfectamente que la falta de homeostasis de la activación del sistema del complemento, en este caso particular debido a la presencia de C3NeF, origina una producción masiva a nivel sistémico de fragmentos de C3 (sobre todo C3b) que se depositan en el glomérulo, causando un daño que puede conducir a insuficiencia renal crónica. Por su parte, cuando se describió la lipodistrofia parcial adquirida apenas se conocía la función clave del sistema del complemento en la biología del tejido adiposo. Se sabe que este sistema es muy importante en la maduración del adipocito, tanto que el tejido adiposo es prácticamente el único productor de FD en el organismo. Sin embargo, aunque existen numerosas evidencias que demuestran el beneficio de esta relación, hay datos que prueban que la desregulación de la vía alternativa se relaciona con la pérdida de tejido adiposo, pero no se conoce con exactitud las causas por el cual este suceso se desarrolla.
El tratamiento más recomendable es el trasplante autólogo de tejido adiposo de zonas no afectadas para restaurar en la mayor parte los problemas de aspecto físico. Es crítico llevar a cabo esta intervención en las etapas más tempranas, sobre todo durante la infancia, para mitigar los trastornos psicológicos asociados. Por otra parte el tratamiento anticomplemento en estos pacientes ha mostrado efectividad bloqueando los efectos dañinos del sistema del complemento en el riñón; pero sin mejoría en lo que respecta al tejido adiposo.
Conceptos clave- •
La lipodistrofia parcial adquirida (síndrome de Barraquer-Simons) es una enfermedad ultra-rara que se caracteriza por la pérdida progresiva del tejido adiposo subcutáneo de la cabeza, el cuello, el tronco y las extremidades superiores, sin afectar a las extremidades inferiores.
- •
La mayor parte de los pacientes con lipodistrofia parcial adquirida presentan niveles disminuidos del componente C3 de la vía alternativa del complemento vinculado con la presencia de C3NeF.
- •
Un 25% de los pacientes desarrollan a medio-largo plazo una C3G, la mayoría por enfermedad por depósitos densos.
- •
La terapia anticomplemento no revierte la pérdida del tejido adiposo, aunque es valorable su empleo en los casos donde se desarrolle una C3G.
Este trabajo ha sido financiado por el Fondo de Investigación Sanitaria del Instituto de Salud Carlos III (PI15-00255), la Sociedad Española de Nefrología (Ayuda para la Investigación de la Fundación SENEFRO, 2016) y CIBERER (ACCI-2015).
Conflicto de interesesLos autores declaran que no tienen conflictos de intereses potenciales relacionados con los contenidos de este artículo.



