






A 58-year-old man with end-stage renal disease (ESRD) secondary to an IgA nephropathy received his first deceased-donor kidney transplant in 2005 and a second one in 2009. Induction immunosuppression consisted of thymoglobulin, mycophenolate, tacrolimus, and corticosteroid. Maintenance immunosuppression consisted of tacrolimus, mycophenolate, and prednisone, with a late switch from mycophenolate to everolimus due to viral infections.
In September 2018, he attended the Emergency Department presenting fever and hypotension, physical examination was normal with no evidence of lymphadenopathy. The serum creatinine was 1.9mg/dl (baseline 1.4mg/dl). The hemogram revealed a high white blood cell count of 10.7×103/μl, with 1.97×103/μl lymphocytes, and hemoglobin of 13.9g/dl.
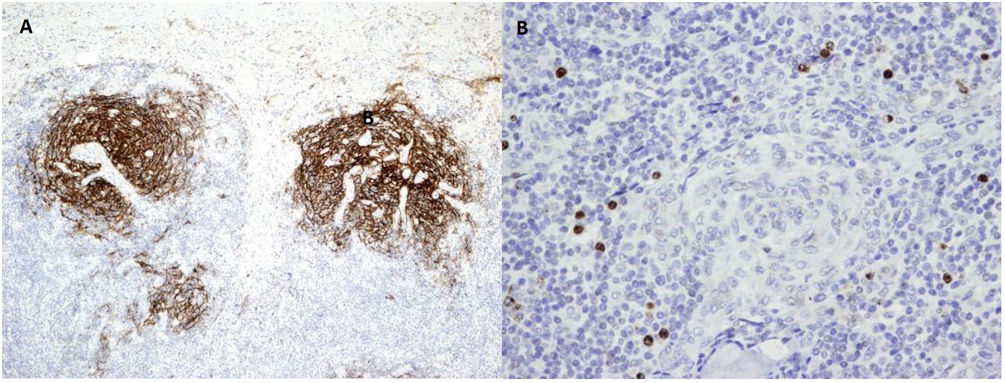
Due to the suspicion of urinary sepsis, cultures (urine and blood) were taken, and he was treated with antibiotics. Despite the use of an appropriate antibiotic, the patient persisted with fever, therefore we decided to request a FDG-PET/CT and polymerase chain reaction (PCR) for various viruses. PCR for CMV, BK, HSV-1, HSV-2, and HV-6 were negatives, while PCR for herpes virus-8 (HV-8) was positive. The result of FDG- PET/CT was the presence of multiple hypermetabolic lymph nodes at both sides of the diaphragm. With the suspicion of a lymphoproliferative syndrome, we reduced immunosuppressive therapy to prednisone and 2 monthly doses of intravenous (IV) immunoglobulins. The histological examination of a lymph node (Figs. 1 and 2) revealed the presence of multicentric Castleman disease (CD) with positivity for HV-8 and after that, the patient was evaluated by the Hematology team. They decided to treat according to the following regimen: valganciclovir for 3 months and four doses of rituximab (4 doses for a month, with a total accumulated dose of 2.8g).
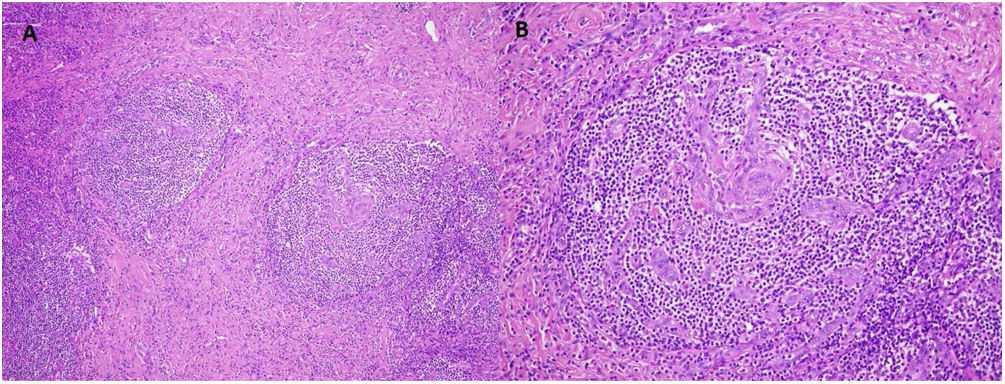
(A) Multicentric Castleman's disease. Follicles with lymphocytes arranged in concentric layers (“onion-skinning”). H/E 10×. (B) Atretic follicular centers with sclerotic blood vessels that radially penetrate the germinal centers, forming hyaline vascular lesions (“lollipop lesions”). H/E 20×.
At present, the patient has no signs of Hematological activity, and renal function keeps stable (mean serum creatinine 1.5mg/dl) without proteinuria. Because of the reduction of immunosuppressive therapy, he developed transient de novo class I donor-specific antibodies. However, he did not exhibit other signs of rejection. Current immunosuppression consists of everolimus, prednisone, and monthly IV immunoglobulins (20g).
DiscussionCD is an uncommon lymphoproliferative disorder that is divided into unicentric or multicentric. It is a condition related to states of immunosuppression, mainly HIV and other viruses such as HV-8.
Although the pathophysiology is not completely understood, one possible explanation involves the proliferation of B lymphocytes that are influenced by levels of interleukin 6 (IL-6) and other molecules. In CD IL-6 increases in a great amount with the consequent proliferation of B-lymphocytes and secretion of immunoglobulin G. In the same way, VEGF (vascular endothelial growth factor) could be secreted in response to IL-61, and in turn, VEGF would contribute to the production of IL-6 by endothelial cells 2.
In the literature, only ten cases of CD related to solid organ transplantation (SOT) have been reported until now. Six of them were related to kidney transplantation 3. In our center, from 1950 to 2020 we have had 24 cases, and only one (the present case) related to a kidney transplant.
Treatment strategies in CD include dose adjustments of immunosuppressive agents. Moreover, higher doses of corticosteroids could reduce lymphocyte proliferation, while a switch from calcineurin inhibitors to sirolimus may promote remissions4, this could be related to lower levels of VEGF and to a lesser viral replication of HV-85. Secondly, patients with an active HV-8 infection should be treated with antivirals6. Additionally, HIV patients should receive antiretroviral treatment. Thirdly, rituximab is the treatment of choice in MCD associated with HV-8 infection. Since the introduction of this drug, survival has dramatically improved from 42% to 94% at two years7. Anti-interleukin-6 receptor monoclonal blockade with tocilizumab could be another option, it is especially interesting because of its potential role in preventing graft rejection8.
In our case, we stopped tacrolimus and we maintained everolimus. As a part of our protocol of reduced IS we also used intravenous pulses of immunoglobulins every month because of its demonstrated antiviral effect on some herpesvirus9 while the patient was treated by the Hematology Department.
We think that the development of de novo DSA could be related to the decrease in IS, and their subsequent decrease could be explained in part by the addition of Rituximab. Comparatively, with the 6 previously reported cases of CD in kidney transplantation, 3 patients died, 1 lost the graft, and 2 survived. Only one of the survivors received chemotherapy (cyclophosphamide, vincristine, doxorubicin, and prednisone), and the other survivor was treated with radiotherapy and a regimen of less intense IS. The other four patients did not receive any special treatment or just a decrease in IS3.
In conclusion, CD is a very uncommon disease, especially in kidney transplant patients. We report the first case of kidney transplant with CD treated with Rituximab, which has proved to be a safe and effective therapy for CD and graft survival.



