












Monoclonal gammopathy of renal significance is a clinical–pathological entity grouping renal disorders secondary to the secretion of a monoclonal immunoglobulin synthesized by a B-cell-derived clone and/or plasma cells in a patient with no diagnostic criteria for multiple myeloma. This term applies to a concept recently introduced owing to the need to differentiate this entity from monoclonal gammopathy of undetermined significance, given the negative prognostic impact of its high morbidity and mortality resulting from both renal and systemic involvement, occasionally even progressing to advanced chronic kidney disease. The renal damage occurs via both direct pathogenic mechanisms, with the deposition of the monoclonal protein in different renal structures, as well as indirect mechanisms, acting as an autoantibody provoking dysregulation of the alternative complement pathway. The detection of this monoclonal protein and an early hematologic study are essential, as is the need for a kidney biopsy to establish the associated nephropathological diagnosis. Consequently, this then leads to the start of specific hematologic treatment to detain the production of the monoclonal protein and minimize renal and systemic injury.
La gammapatía monoclonal de significado renal es una entidad clínico-patológica que agrupa los trastornos renales secundarios a la secreción de una inmunoglobulina monoclonal sintetizada por un clon derivado de células B y/o células plasmáticas en un paciente sin criterios de diagnóstico de mieloma múltiple. Este término se aplica a un concepto introducido recientemente debido a la necesidad de diferenciar esta entidad de la gammapatía monoclonal de significado incierto, teniendo en cuenta el impacto pronóstico negativo de su alta morbilidad y mortalidad a causa de la afectación tanto renal como sistémica, llegando en ocasiones a progresar a una enfermedad renal crónica avanzada. El daño renal se produce tanto por mecanismos patogénicos directos, con el depósito de la proteína monoclonal en diferentes estructuras renales, como por mecanismos indirectos, actuando como un autoanticuerpo que provoca la desregulación de la vía alternativa del complemento. La detección de esta proteína monoclonal y un estudio hematológico precoz son imprescindibles, así como la necesidad de una biopsia renal para establecer el diagnóstico nefropatológico asociado. En consecuencia, esto lleva al inicio de un tratamiento hematológico específico para detener la síntesis de la proteína monoclonal y minimizar la lesión renal y sistémica.
Although kidney disease is a common complication of malignant hematologic cancers, most kidney diseases are associated with nonmalignant blood dyscrasias, such as monoclonal gammopathy (MG). Monoclonal gammopathy of uncertain significance (MGUS) is the most usual form. It is more frequent at older ages, affecting about 3% of the population over 50 years of age. It is defined as a clonal proliferative disorder of B cells or plasma cells, producing a monoclonal immunoglobulin with a monoclonal peak below 30g/L, infiltration of anomalous plasma cells into the bone marrow less than 10% and absence of clinical evidence of myeloma, lymphoma or amyloidosis. A lesion is considered to be nonmalignant or premalignant if it does not damage any target organ. The monoclonal immunoglobulins are mainly of the classes IgG, IgA or IgM.
The term monoclonal gammopathy of renal significance (MGRS) was proposed by the International Kidney and Monoclonal Gammopathy Research Group (IKMG) in 2012. It describes those disorders of MGUS with renal involvement associated with a monoclonal protein. The IKMG later redefined MGRS as a clonal proliferative disorder of B cells or plasma cells that produces a monoclonal immunoglobulin with nephrotoxic effects and that does not fulfill the hematologic criteria to start the treatment of a specific malignant cancer.1 This monoclonal protein may be light chain, heavy chain or an intact immunoglobulin.
The reason for defining this new entity arose from the need to differentiate patients with MG but with no lesion in any other organ from patients at risk of developing kidney disease, as these latter have a high degree of morbidity and mortality due to the severity of their kidney (and sometimes systemic) lesions induced by the monoclonal protein. Early recognition and characterization of the kidney lesion is crucial in order to start treatment and detain the secretion of the paraprotein, thereby avoiding progression to end-stage renal disease and improving both kidney and patient survival.
Monoclonal gammopathy is considered a nonmalignant hematologic disease and does not require any specific hematologic treatment. Its effects on the kidney are sometimes deleterious, as reflected in a retrospective study that included 19 patients with light-chain deposition kidney disease, of whom just 12 had MGUS. After a follow-up of five years kidney and patient survival were 37% and 70%, respectively.2 Steiner et al. showed that patients with MGRS had a greater risk of progression to multiple myeloma (MM) than those with MGUS, with a mean progression time of 18.8 years.3
Clinical manifestationsMonoclonal gammopathy of renal significance can present with a wide variety of clinical symptoms. The toxicity of the monoclonal protein is not just limited to the kidney; it can also cause neuropathy, dermopathy, eye disorders and other extrarenal conditions. The kidney is the organ most affected in dysproteinemias, as several physiological factors come together to increase the potential risk of damage. After the lung, the kidney is the organ exposed to the greatest cardiac output. The kidney is also immersed in a medium where the pH and the electrolyte concentrations can alter the physical and chemical characteristics of the monoclonal protein, making it more toxic.
Renal involvement is generally manifested by varying degrees of proteinuria, which can reach the nephrotic range, or it may also be associated with microhematuria and hypertension. A high percentage of patients have kidney failure at the time of diagnosis, which can progress to end-stage renal disease.4 Proteinuria with albuminuria is the most common finding if there is glomerular involvement. However, if there is tubulointerstitial involvement, as occurs in Fanconi syndrome, it is necessary to assess proximal tubular dysfunction, noting in these cases the presence of glycosuria, uricosuria, phosphaturia and proximal renal tubular acidosis.
Extrarenal manifestations are not uncommon. In light-chain amyloidosis, in addition to renal damage, there can also be cardiac, hepatic or neurologic involvement. In cryoglobulinemic glomerulonephritis joint and skin involvement is common. MGRS can also indirectly cause lesions to the vascular endothelium via deregulation of the alternative complement pathway or the release of endothelial growth factors. In these cases the disease can present with cutaneous lesions, thrombotic microangiopathy and the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, MG, and skin changes).
Diagnosis: hematologic evaluation and renal evaluationHematologic evaluationIn order to establish the hematologic diagnosis of MG it is necessary to follow the diagnostic criteria and the classification of the four clinical entities that are grouped under this term, as established by the International Myeloma Working Group in 2014.5
Immunoglobulins are composed of two identical heavy chains, bound to two identical free light chains (FLC), either kappa or lambda. The plasma cells produce an excess of FLC, which is secreted simultaneously with the immunoglobulins intact to the blood. Forty percent of FLC are found in their free form in serum and not attached to heavy chains, and their amount in serum depends on both the level of production by plasma cells and the rate of renal metabolization. The glomerulus filters molecules with a cut-off point between 40 and 60kDa, with a FLC kappa size of 25kDa and lambda size of 50kDa. Thus, both are filtered by the glomerulus and then reabsorbed and metabolized in the proximal tubule. The capacity of reabsorption of the FLC by the kidney is 10–30g/day. Therefore, under normal physiological conditions, between 0.5 and 1g/day of FLC will be reabsorbed in their entirety at the renal level. However, if the circulating levels of serum FLC are increased due to higher production, the capacity for renal resorption and metabolization will be saturated and from this point onwards Bence Jones proteinuria will be seen, with FLC detectable in urine. This saturation is associated with an increased risk of nephropathy and deterioration of renal function. As the glomerular filtrate decreases, fewer FLC will be filtered and the 24-h urine FLC evaluation would cease to be useful as a diagnostic process, as it no longer reflects the clinical hematologic condition of the patient.6
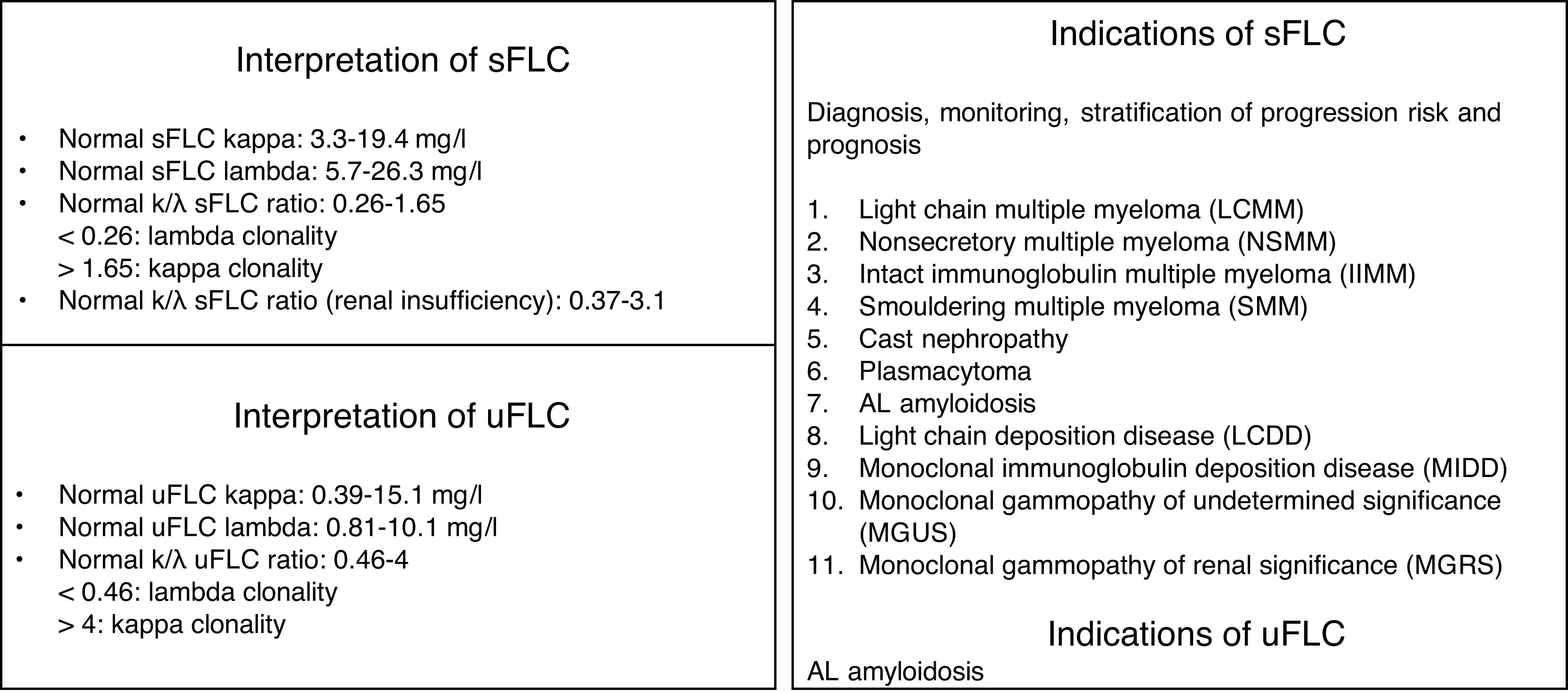
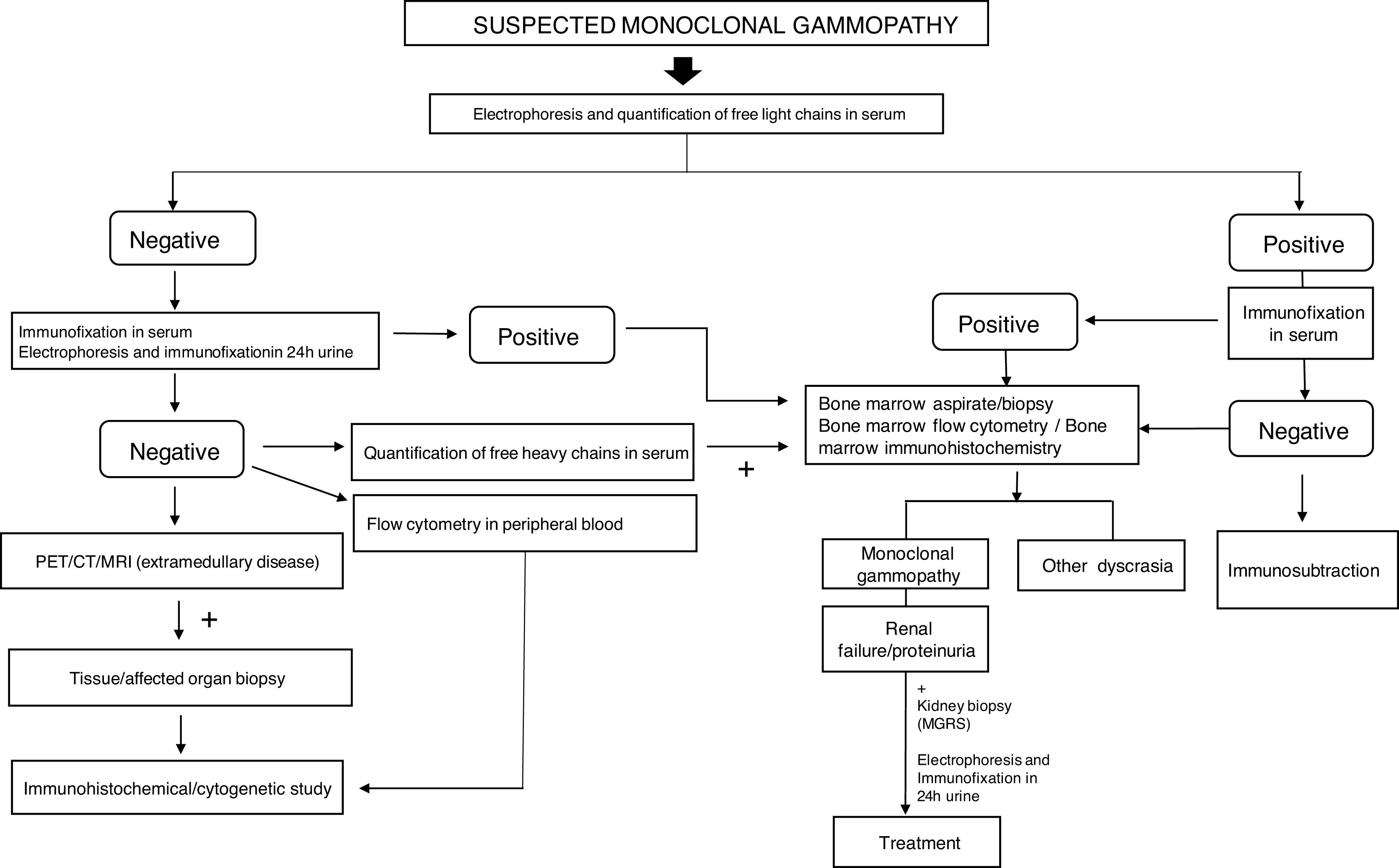
For the evaluation of monoclonal protein, a comprehensive hematologic study should be performed that should include a serum study, 24-h urine, and bone marrow biopsy/aspiration. Serum protein electrophoresis is the most widely used technique as it allows detection of existing monoclonal bands, but its sensitivity is limited because it cannot detect monoclonal peaks lower than 400mg/L. Immunofixation is a more sensitive technique that allows the characterization of the type of immunoglobulin but not its quantification. Quantification of FLC is done by serum FLC assay, which is the most sensitive technique. It enables the measurement of normal serum FLC levels and the determination of reference ranges, with the normal range for kappa FLC from 3.3 to 19.4mg/L and for lambda FLC from 5.7 to 26.3mg/L, with a normal kappa/lambda ratio between 0.26 and 1.65. This kappa/lambda ratio may be altered with kidney failure. Hutchison et al. established a normal range in patients with kidney failure of 0.37 to 3.1, considering it unlikely that a kappa/lambda ratio greater than 3 could be due to the presence of kidney failure.7 Monoclonal protein can be identified in 25–76% of cases by serum electrophoresis and immunofixation. In cases that are not identified with these techniques, the serum FLC assay should be used.8 The serum FLC assay has great diagnostic use to identify the monoclonal paraprotein in those cases where it is not detected by immunoelectrophoresis, in addition to being important for the follow-up, monitoring and prognosis of the condition (Fig. 1). Occasionally, however, the clone is small and unable to be detected with immunoelectrophoresis. This therefore makes it difficult to detect the clonality for the diagnosis. Consequently, we propose an algorithm for its identification.9,1,10 (Fig. 2).
Bone marrow aspiration and/or biopsy should be performed and should include immunohistochemistry and flow cytometry to determine cell surface and intracellular markers in plasma cells and B cells. Immunohistochemistry staining for CD138 in bone marrow biopsies is necessary to adequately quantify plasma cells, with clonal B cell populations characterized by their positivity for CD5, CD10 and CD20 markers.11
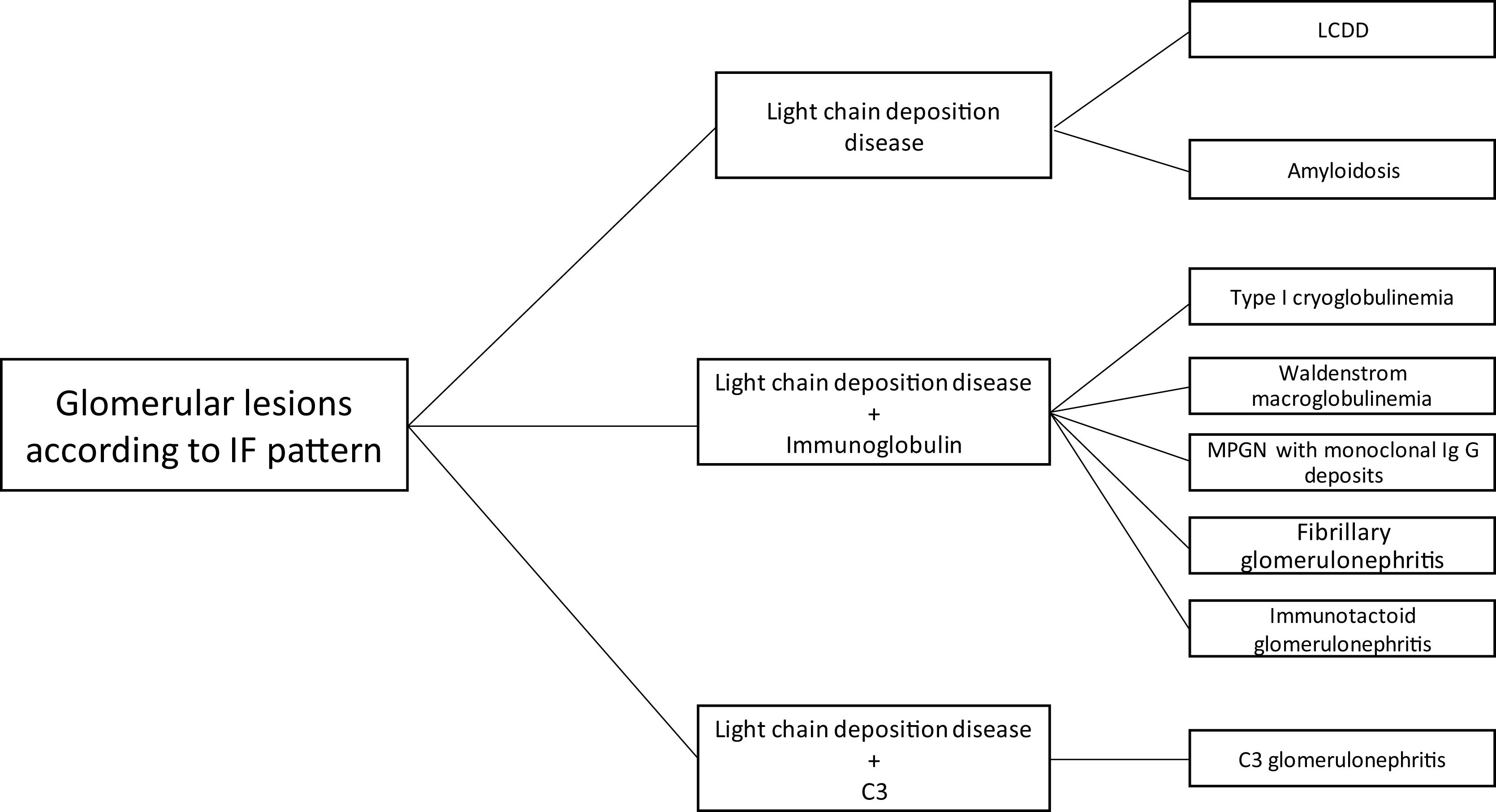
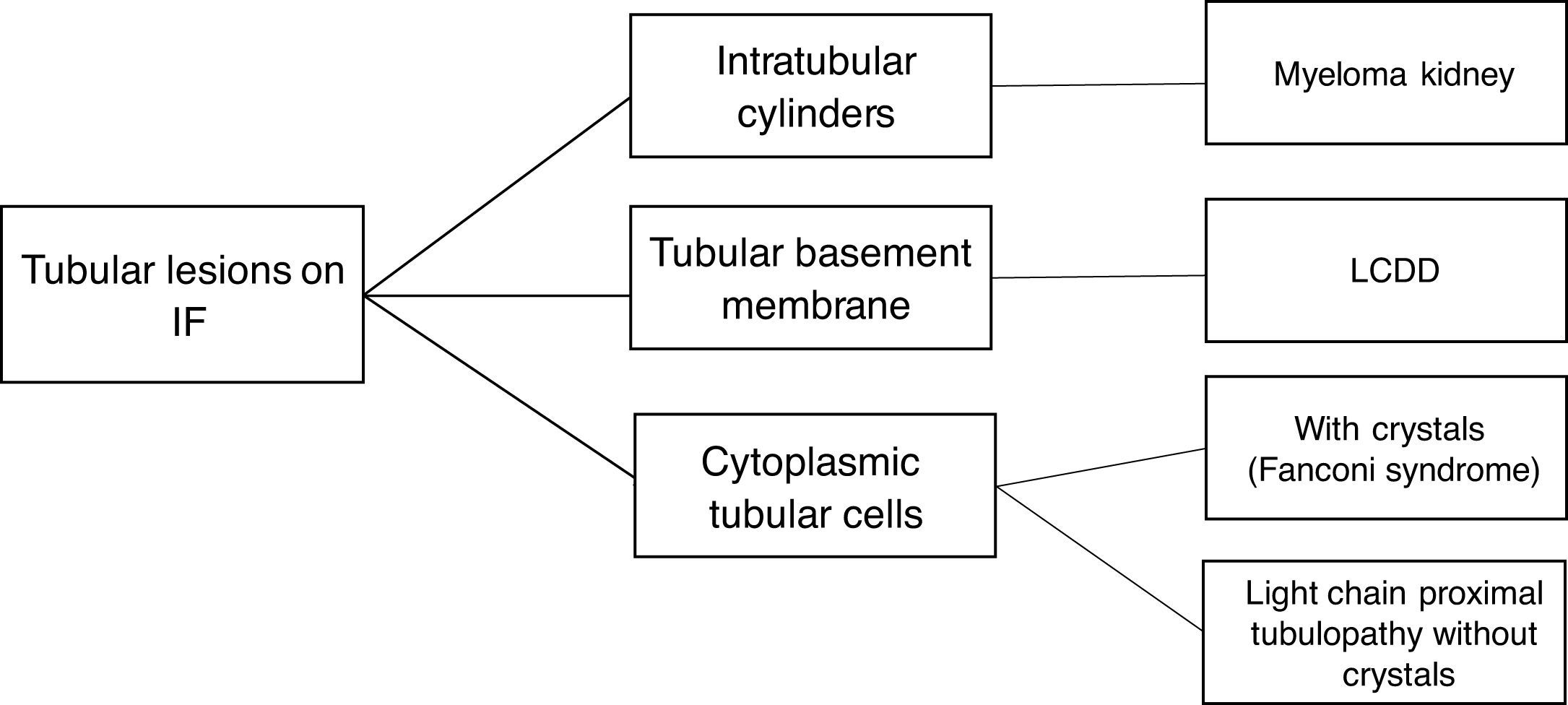
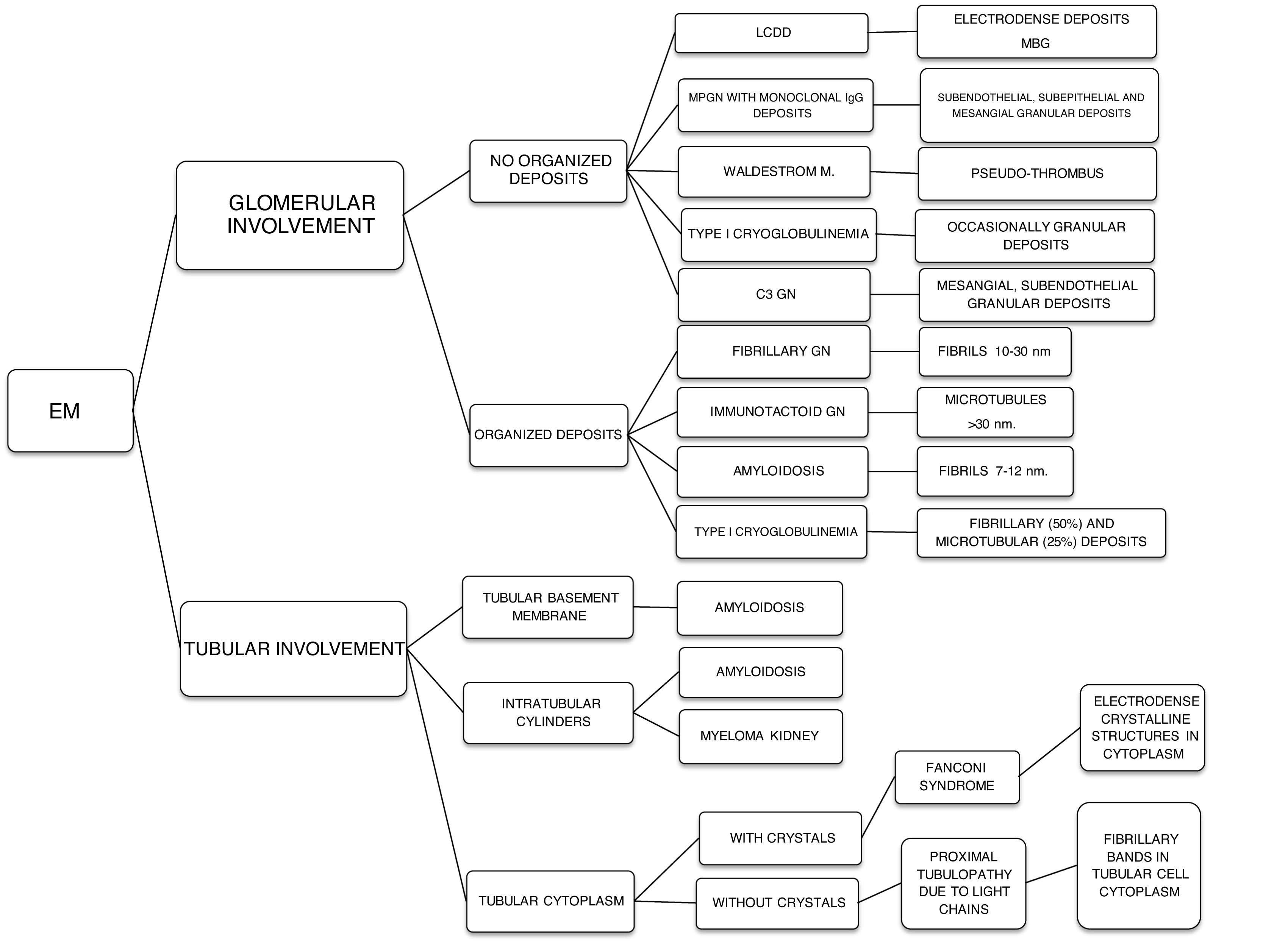
The hematologic study of a dysproteinemia is often started after performing a kidney biopsy due to worsening renal function. Study of the biopsy with immunofluorescence shows the deposit of a monoclonal immunoglobulin or C3. With these findings we should discard the presence of MG and start the hematologic study to identify the monoclonal protein using immunofixation and electrophoresis in serum and in 24-h urine, as well as perform a bone marrow biopsy. In those cases where renal biopsy shows a predominant deposit of C3 with absence of monoclonal immunoglobulin it is necessary to perform a genetic study and functional complement testing, as well as a hematologic study of the dysproteinemia, as the monoclonal protein may cause a change in the dysregulation of the alternative complement pathway and its renal deposits (Figs. 3–6).
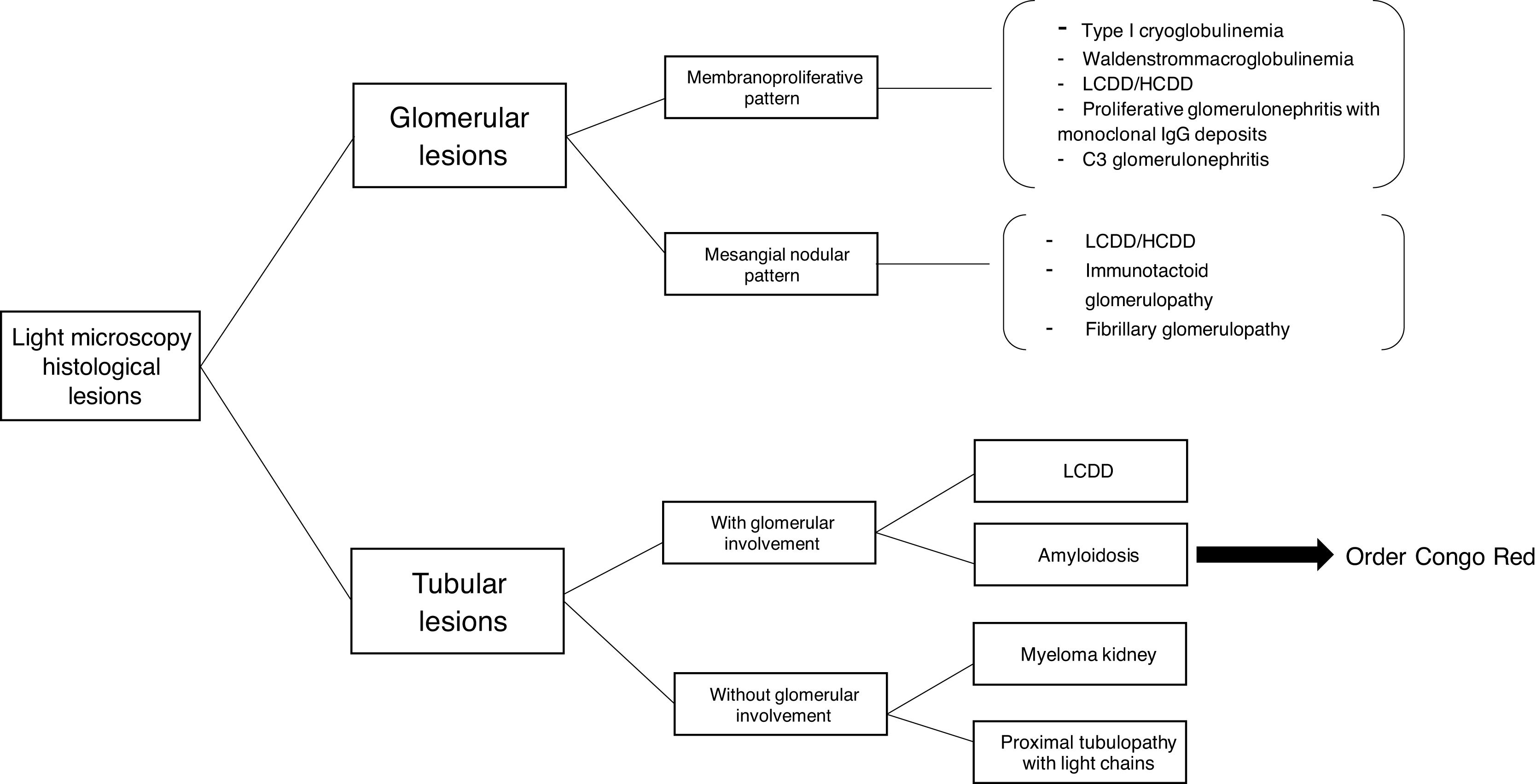
Depending on the tumor load, two main categories of kidney disorders associated with dysproteinemias should be distinguished. The first group requires the secretion of large amounts of monoclonal immunoglobulin, precipitating into the lumen of the distal renal tubule, characteristic of cast nephropathy, which is the most common cause of renal disease in MM. The second group of kidney diseases is related to secretion with a low tumor load, which is what usually occurs in MG and is detailed below.
The spectrum of kidney diseases associated with MG is so broad that several histopathological lesions may coexist in the same patient. Several mechanisms of nephrotoxicity have been described that can affect different renal compartments (glomeruli, vessels, tubules and interstitium). These renal injury mechanisms include deposition, precipitation, complement activation and cytokine secretion (Fig. 7). Deposition is the most common mechanism and is observed in amyloidosis, monoclonal immunoglobulin deposition disease (MIDD), membranoproliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID), immunotactoid glomerulonephritis and fibrillary glomerulonephritis. These deposits may be organized, as in amyloidosis, fibrillary and immunotactoid glomerulonephritis, or nonorganized, as in MIDD and PGNMID. Precipitation occurs in cast nephropathy, cryoglobulinemic glomerulonephritis, and crystal-induced nephropathy. Intravascular precipitation occurs in the glomerular capillaries in cryoglobulinemia and tubular precipitation is seen in cast nephropathy.12 Complement activation and cytokine secretion are observed in C3 glomerulopathy and POEMS syndrome, respectively.
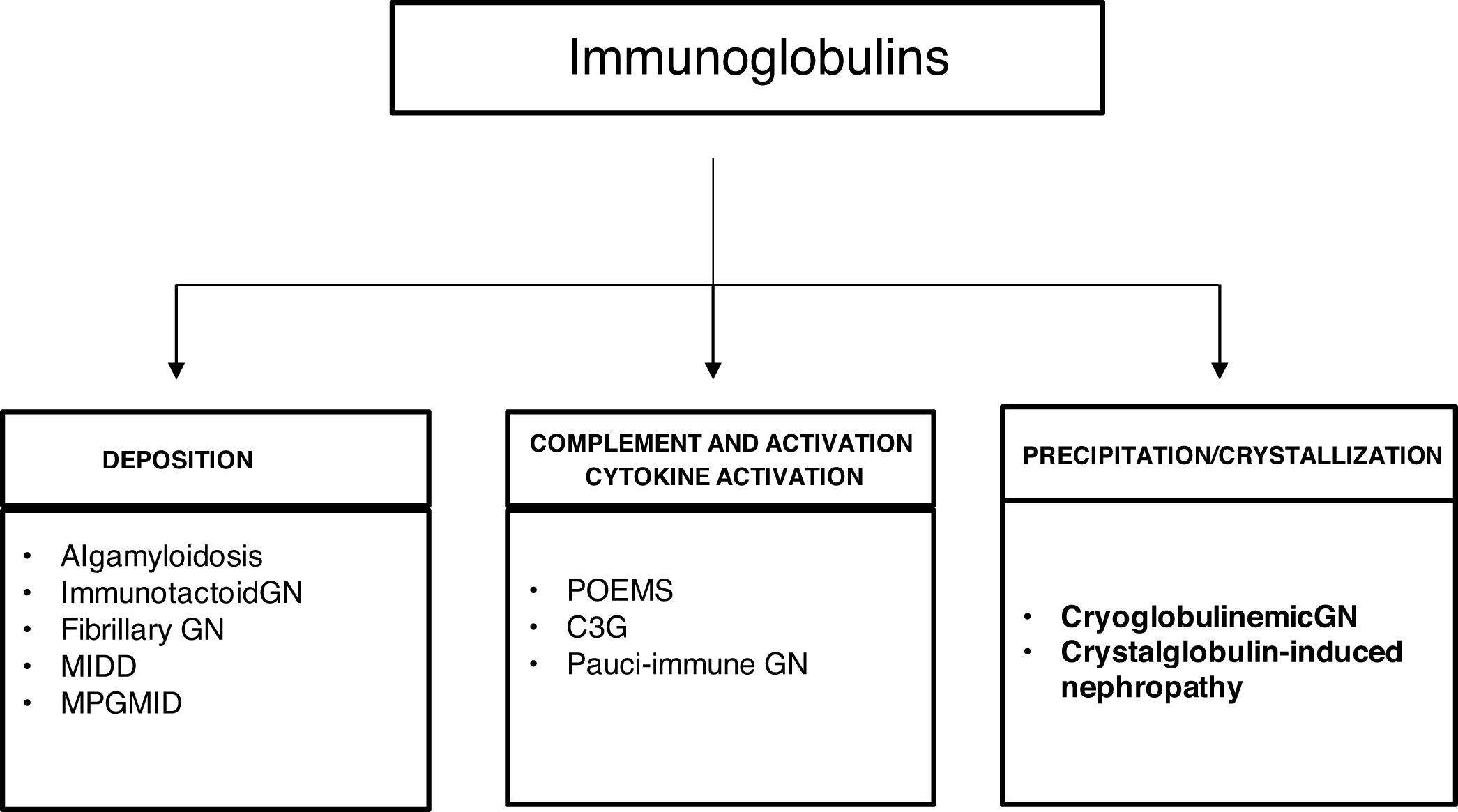
Mechanisms of glomerular toxicity. AIg amyloidosis (immunoglobulin-derived amyloidosis), MIDD (monoclonal Ig deposition disease), MPGMID (proliferative GN with monoclonal Ig deposits), POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes), C3G (C3 glomerulopathy).
Activation of the alternative complement pathway causes C3 glomerulopathy in the kidneys, although the nephritic factor and anti-factor H autoantibody are present in some patients. Others present genetic polymorphisms of the complement that predispose to this entity. However, in most cases the mechanism by which the monoclonal protein can cause deregulation of the alternative complement pathway is unknown, the main hypothesis being that this protein may act as an autoantibody directed against complement regulatory factors, triggering an activation of the alternative complement pathway.13
Other mechanisms of renal injury involve the secretion of various biological factors and/or the activity of circulating autoantibodies by the monoclonal protein that can target the A2 phospholipase receptor causing membranous nephropathy, or against the glomerular basement membrane causing Goodpasture syndrome.14 In addition, cytokines can be activated and high levels of vascular endothelial growth factor secreted, which are implicated in the risk mechanism for the development of thrombotic microangiopathy or POEMS syndrome nephropathy.15 Eight percent of patients with light chain amyloidosis and 20% of those with MIDD present symptomatic MM at the time of diagnosis.8
Renal biopsy is necessary to determine the histopathology associated with MGRS and to evaluate its severity by demonstrating monoclonal deposits in the kidney. It is indicated in patients with kidney failure and/or significant proteinuria, as well as in those patients with active urinary sediment.16 Lin et al., studied patients with MIDD and observed that up to 39% presented MGUS and the renal biopsy findings preceded the diagnosis of dysproteinemia in 68% of cases.17 A detailed study should be performed to establish the equivalence of monoclonal protein in blood circulation and in the kidneys.
No useful biomarkers exist for the early detection of kidney damage in MM or MG. Iriuchishima et al. studied the role of activin A, a cytokine from the TGF-β (transforming growth factor beta) superfamily that is involved in the development of certain kidney diseases. These authors analyzed the levels of activin A in the urine of patients recently diagnosed with MM (n=41), latent MM (n=10) and MGUS (n=28), including MGRS, finding that this cytokine correlated significantly with the level of serum and urinary monoclonal protein, but was absent in renal biopsy samples without associated renal disease. It was, however, detected in the tubular cells of patients with MGRS. Therefore, these authors suggest that activin A is a urinary biomarker that reflects renal tubular damage in MM and MG and that may aid in the early detection of nephrotoxicity in plasma cell dyscrasias, although it does not establish the associated renal disorder documented by renal biopsy.18
Currently, histopathological renal lesions are classified in two groups according to the affected renal compartment: tubular or glomerular disorder.8 However, amyloidosis associated with immunoglobulin and MIDD may present a mixed pattern of glomerular and tubular involvement.
Tubular disorders in MGRSThe most frequent alterations as a consequence of tubular dysfunction are: hypouricemia, hypophosphatemia, normoglycemic glycosuria, aminoaciduria, proximal tubular acidosis, and tubular proteinuria (without albuminuria). Progressive kidney failure is also seen. Among the main tubular disease entities associated with MGRS are the following:
- 1.
Light chain-associated Fanconi Syndrome presents the characteristic alterations of tubular dysfunction, as well as tubular proteinuria and slow evolution to end-stage kidney failure. Renal biopsy shows atrophy and dedifferentiation of proximal tubular cells (PTC) with intracytoplasmic inclusions. On immunofluorescence, light chains are deposited in PTC. In 90.9% of cases these are kappa FLC.19
- 2.
Proximal tubulopathy without crystals slowly evolves to end-stage kidney failure with the presence of tubular proteinuria. On light microscopy and immunofluorescence we observe similarities with Fanconi syndrome, but on immunofluorescence both kappa and lambda light chains may be seen.
- 3.
Crystal-storing histiocytosis presents crystal deposits formed by light chains in the PTC that may also appear in the lysosomes of the histiocytes in the bone marrow. It presents the characteristic alterations of tubular dysfunction with slow impairment of renal function. On light microscopy, histiocytes with crystalline inclusions in the renal interstitium (pseudo-Gacher cells) are visualized with atrophy and dedifferentiation of PTC in the vicinity of perirenal fat. Immunofluorescence shows mainly kappa FLC inclusions in the PTC.
- 4.
Cast nephropathy is associated with MM in 90% of cases and is less frequent in MG, which requires a high tumor load.
The most common clinical manifestations are proteinuria, microhematuria, nephritic or nephrotic syndrome, hypertension and kidney failure, generally related to the main associated pathologies listed below:
- 1.
Immunoglobulin light chain-related amyloidosis is produced by the secretion of kappa or lambda FLC by the B cell clone. Involvement of a heavy chain is extremely rare. The fibrils in light chain amyloidosis are derived from the variable region of the lambda FLC in 75% of cases. This type of amyloidosis is associated with elevated proteinuria and nephrotic syndrome, and kidney failure is usually present at diagnosis in 70–80% of cases.20 On light microscopy, glomeruli show massive amyloid deposits (eosinophilic, pale and acellular material), but other renal compartments such as arterioles, arteries and the tubulo-interstitial compartment may also show these deposits. On immunofluorescence, lambda light chain staining is characteristic, and on electron microscopy amyloid is identified as unbranched fibrils with a diameter of 8–10nm. The definitive diagnosis is made by Congo red staining that detects apple green birefringence under polarized light in amyloid deposits.
- 2.
Fibrillary glomerulonephritis is rarely a primary glomerular disease. Fifty percent of cases present proteinuria in the nephrotic range, with or without kidney failure, hypertension or microhematuria. Fibrillary deposits have a larger diameter than amyloid deposits and are Congo red negative. Light microscopy shows a pattern of membranoproliferative glomerulonephritis (MPGN) and mesangial proliferation with fibrils deposited in the mesangium and/or glomerular basement membranes. Immunofluorescence highlights polyclonal glomerular IgG and FLC deposits are more frequent than monoclonal deposits. Electron microscopy shows randomly aligned fibrils.12
- 3.
Immunotactoid glomerulonephritis presents clinically with nephrotic syndrome, with infrequent extrarenal involvement and microtubular immunoglobulin deposits organized at the glomerular level that on electron microscopy measure more than 30nm in diameter and are negative for Congo red. On light microscopy, an MPGN or membranous pattern is observed on immunofluorescence staining for monoclonal IgG and complement components.
- 4.
Cryoglobulinemic glomerulonephritis usually presents with nephritic syndrome and kidney failure that progress in flare-ups, with frequent extrarenal, cutaneous, articular and neurologic involvement. There are three types of cryoglobulinemia. Type I is associated with a monoclonal immunoglobulin generally of the IgG or IgM class, with more frequent renal manifestations when associated with IgG20 and is associated with lymphoproliferative disorders. Types II and III are mixed cryoglobulinemias, frequently associated with infections and autoimmune disorders, respectively, that present with immunocomplexes formed by monoclonal and polyclonal immunoglobulin in Type II and immunocomplexes formed by polyclonal immunoglobulins in Type III. In mixed cryoglobulinemias, the elevation of rheumatoid factor and serum complement anomalies are more frequent than in Type I. On light microscopy, an MPGN pattern is seen with immunofluorescence staining for IgG, IgM and complement components. Electron microscopy shows subendothelial and intracapillary organized fibrillary or microtubular deposits.
- 5.
Monoclonal immunoglobulin deposition disease (MIDD) is a rare complication in MG, although when it occurs it is usually associated with extrarenal manifestations with hepatic and cardiac involvement, with renal manifestation of proteinuria in the nephrotic range and kidney failure. MIDD is associated with the deposit of light chain, heavy chain or both light and heavy chain. It is most frequently associated with kappa light chain and when associated with heavy chain, this is usually lambda. Deposits are nonorganized and Congo red staining is negative. Light microscopy shows nodular sclerosing lesions and thickening of tubular basement membranes; a membranoproliferative pattern can also be observed. On immunofluorescence, monoclonal light and heavy chains in glomerular and tubular basement membranes are stained linearly and diffusely. On electron microscopy, dense punctiform deposits are seen in the glomerular and tubular basement membranes.21 Joly et al. retrospectively analyzed a cohort of 255 patients with histological diagnosis of MIDD associated with MG and MM in 64% and 35%, respectively. Approximately 35% of cases had extrarenal involvement, mainly cardiac and hepatic. Of the 169 patients who received treatment (58% with Rituximab), hematologic and renal remission was achieved in 67% and 36%, respectively. All the patients who had a renal response also had a hematologic response, with the absence of severe interstitial fibrosis and a hematologic response being predictive factors of renal response.22
- 6.
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) manifests with proteinuria, microhematuria, hypertension and kidney failure with C3 hypocomplementemia. It is an MPGN mediated by immune complexes in the mesangial and subendothelial areas. A membranoproliferative pattern is observed on light microscopy, with immunofluorescence staining for a single light- or heavy-chain isotype, most commonly IgG3. On electron microscopy, granular nonorganized deposits are seen in the mesangial, subendothelial and intramembranous areas.21
- 7.
C3 glomerulopathy includes two subtypes, C3 glomerulonephritis and dense deposit disease, which can be differentiated under electron microscopy. Both manifest with proteinuria, microhematuria with or without kidney failure and C3 hypocomplementemia. Monoclonal protein is not deposited directly into renal structures, but may interfere with the regulatory proteins of the alternative complement pathway, such as factor H or act as a nephritic factor, resulting in dysregulation of the alternative complement pathway, through abnormal control of its activation, degradation or deposition.23 On light microscopy, several mesangial proliferation, membranoproliferative and endocapillary proliferation patterns can be observed. Immunofluorescence shows intense C3 deposits in the mesangium and capillaries, with absence or scarcity of other reactants. The two subtypes are differentiated on electron microscopy. While C3 glomerulonephritis presents nonorganized dense deposits in the mesangial, intramembranous and subendothelial areas, in dense deposit disease the deposits are only in the mesangial and intramembranous areas.12
The most accepted classification of MGRS-associated lesions is based on the distinction according to the organization of deposits on electron microscopy, whether they are organized (fibrils, microtubules, crystals) or nonorganized24 (Table 1).
Classification of renal disorders associated with monoclonal gammopathy according to the organization of the deposits under electron microscopy.
| Organized deposits | Nonorganized deposits |
|---|---|
| Fibrils:- Monoclonal immunoglobulin-related amyloidosis- Fibrillary glomerulonephritis | Monoclonal immunoglobulin deposition disease (MIDD) |
| Microtubules:- Immunotactoid glomerulonephritis- Type I cryoglobulinemic glomerulonephritis | Proliferative glomerulonephritis with monoclonal immunoglobulin deposits |
| Crystals:- Proximal tubulopathy- Crystal-storing histiocytosis | C3 glomerulopathy |
The goal of treating patients with MGRS is to stop the natural progression of the kidney disease. The time from diagnosis to treatment is of vital importance as prolonging this time increases the likelihood of irreversible renal and systemic complications due to progression to tubulointerstitial fibrosis with development of end-stage kidney failure. Multiple studies show that a rapid decrease in serum FLC allows a high rate of renal recovery.4 The nephrologist has a central role in early diagnosis, since in most cases patients present with kidney failure without a known diagnosis of MG, with the findings on renal biopsy preceding the hematologic diagnosis. Thus, treatment from early stages has a great impact on the prognosis of renal and patient survival.11
It is important to emphasize that kidney transplantation in patients with MGRS without previous treatment may present a significant risk of renal allograft loss due to disease recurrence, since the mechanism of the lesion remains in full activity.25 For this reason, treatment should also be considered in those patients who are candidates to receive a kidney transplant, since in this case complete remission of the hematologic disease may prevent subsequent recurrence in the renal transplant.26
In 2013 the IKMG published a consensus document that includes treatment strategies for MGRS based on the underlying clone and the severity of the disease20,27,28 (Table 2).
Treatment of MGRS proposed by the International Kidney and Monoclonal Gammopathy Working Group 2013: CKD (chronic kidney disease), SCT (stem cell transplantation).
| Monoclonal Ig-related amyloidosis | - Classification in three stages according to heart involvement- Stage I–II: melphalan or cyclophosphamide+dexamethasone+bortezomib. If no response, consider SCT- Stage III: cyclophosphamide+dexamethasone+bortezomib. In select cases SCT. |
| Monoclonal Ig deposition disease (MIDD) | - CKD I–III: cyclophosphamide+dexamethasone+bortezomib. SCT in select cases- CKD IV–V: cyclophosphamide+dexamethasone+bortezomib. If no response and if kidney transplant candidate, SCT. |
| Proliferative glomerulonephritis with monoclonal Ig deposits (PGNMID) | - CKD I–II+proteinuria<1g/day+no evidence of renal progression: observation- CKD I–II+proteinuria>1g/day+evidence of renal progression or CKD III–IV: cyclophosphamide+dexamethasone+bortezomib. Consider rituximab when the B clone is CD20- CKD V+kidney transplant candidate: SCT |
| Type I cryoglobulinemia | - Few systemic symptoms: observation- Systemic and/or progressive disease:1. If plasmocytic clone: bortezomib+dexamethasone+/−thalidomide. In select cases consider SCT2. If lymphoplasmocytic clone: regimens with rituximab.In any case, plasma exchange is done |
| Immunotactoid glomerulonephritis | - Severe CKD: cyclophosphamide and/or bendamustine+steroids+/−rituximab- Isolated monoclonal gammopathy: bortezomib |
| Fanconi syndrome | - CKD I–III: cyclophosphamide+bortezomib or thalidomide. Second line, SCT- CKD IV–V+kidney transplant candidate: SCT- CKD IV–V+not kidney transplant candidate: observation |
The last ten years have seen a revolution in the treatment of MM, with the identification of CD38 and the signaling lymphocytic activation molecule family (SLAMF).29–31 These have enabled the introduction of various molecules whose mechanisms are based on proteasome inhibition, blockade of histone deacetylase (HDAC) and immunomodulation via the monoclonal antibodies daratumumab and elotuzumab, approved by the Food and Drug Agency (FDA) in 2015.30 The HDCA drugs panobinostat and rocilinostat (ACY-1215) are under experimentation. Daratumumab is a monoclonal antibody directed against CD38+ cells. Its use has been approved in combination in cases of refractory and/or recurrent disease, and also as first-line therapy in patients not eligible for transplant of hematopoietic progenitor cells. Elotuzumab is a monoclonal antibody directed against SLAMF7 and is used in combination with lenalidomide and dexamethasone in patients already treated with one or three drugs. Denosumab is another monoclonal antibody approved by the FDA to treat MM-associated bone loss.30,32,33 The era of immunotherapy for MM was opened up by the approval of these two drugs in 2015, with current clinical trials studying other promising molecules for the treatment of MM. When these can be incorporated there will be other new alternative therapies for MM (Fig. 8).
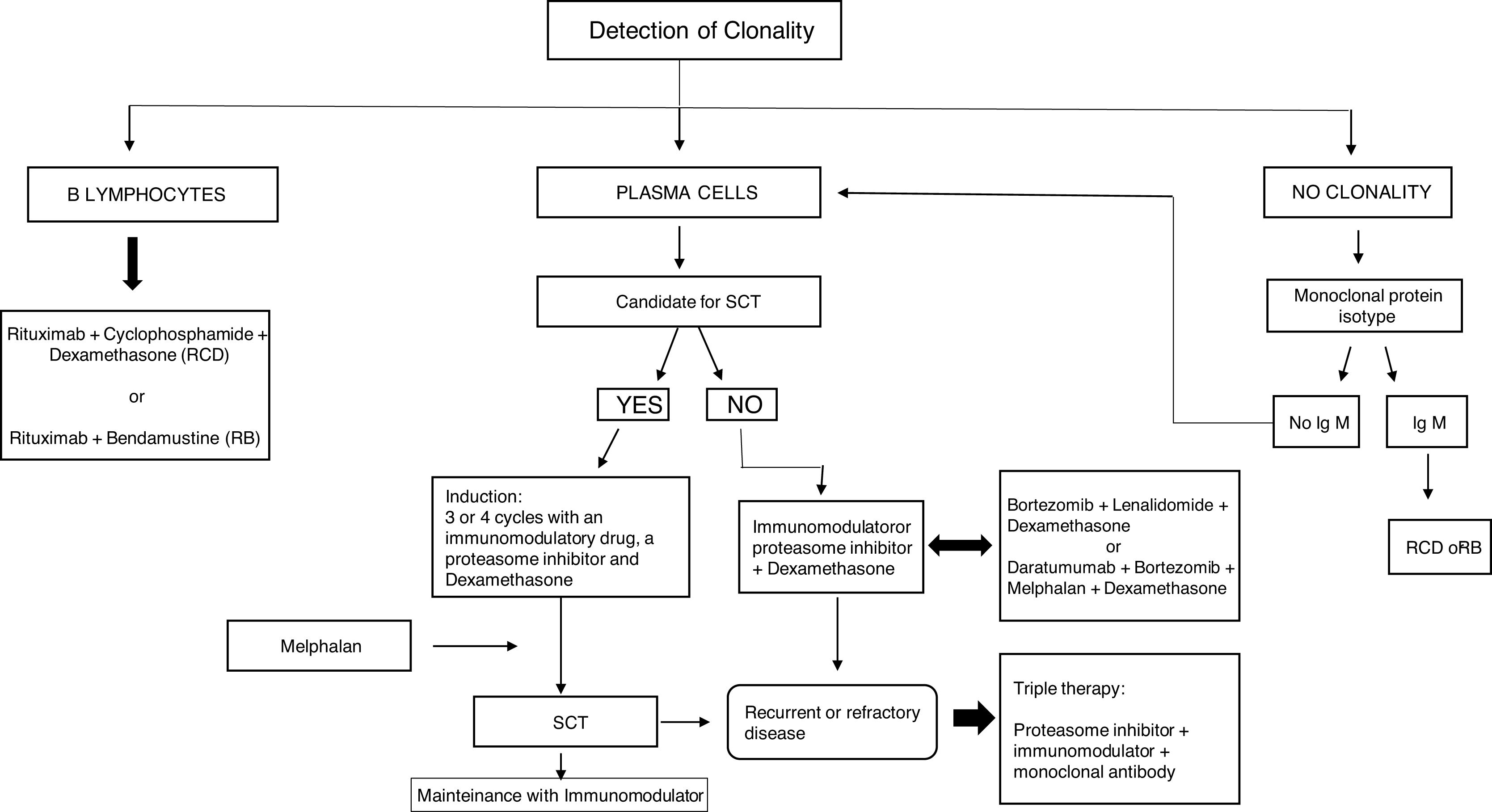
Follow-up of MGRS patients should be close both during treatment and afterwards. It must include clinical and laboratory controls, involving multidisciplinary assessment by hematologists and nephrologists to evaluate the treatment response, as well as any possible treatment-associated complications. The disease is often refractory or recurrent, requiring various lines of treatment (Fig. 9).
Conclusions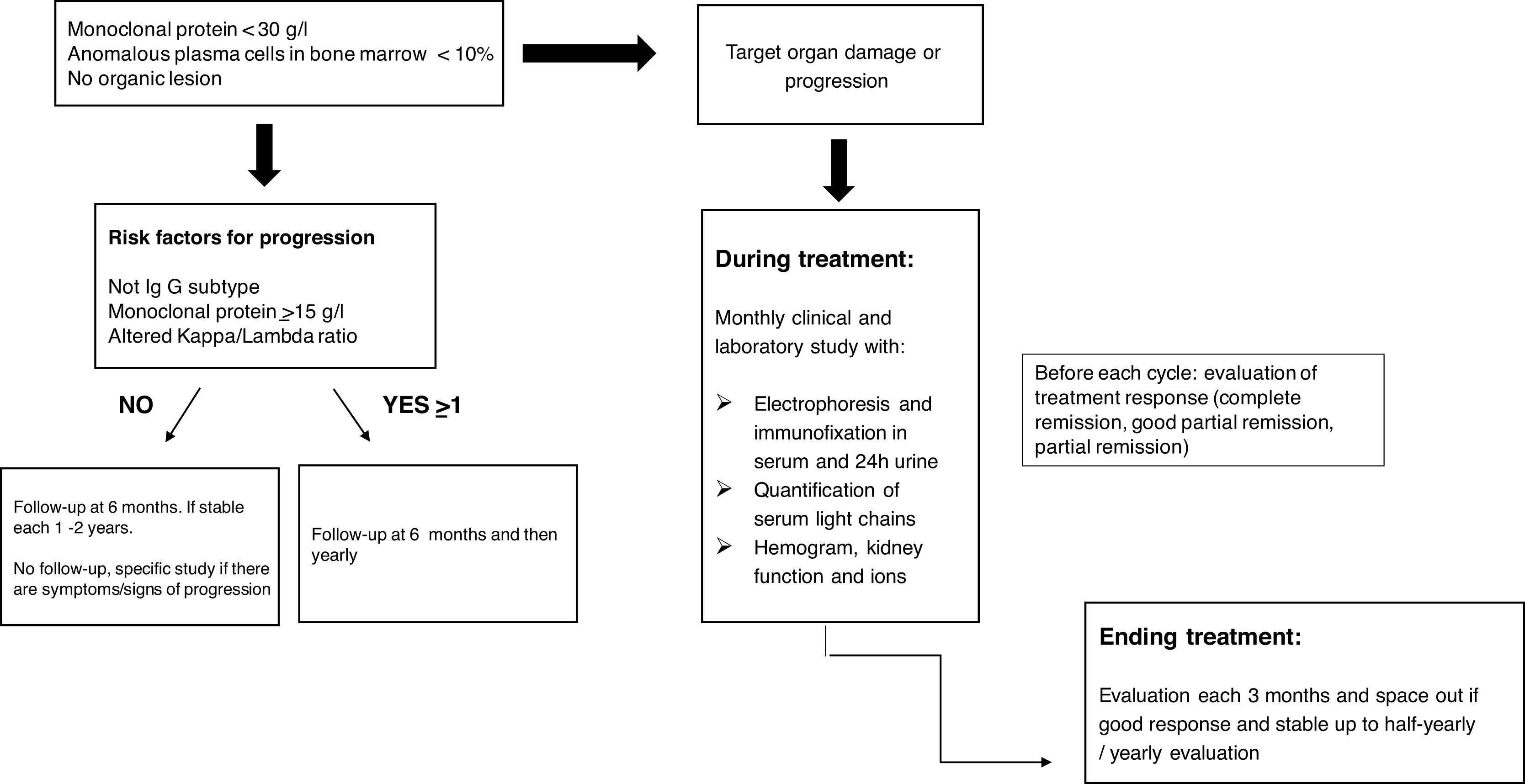
Monoclonal gammopathy of renal significance is a newly diagnosed entity that encompasses a broad spectrum of kidney diseases associated with MG. In an elevated number of cases, the presence of kidney failure and/or alterations in urinary sediment precede the hematologic diagnosis. In many cases, renal histological lesions condition the search for MG or other blood dyscrasia as the underlying cause of kidney failure.
Although MG does not initially require treatment from the tumor standpoint, when there is associated renal disease, treatment should be considered obligatory and early, with the appropriate therapeutic regimen based on the underlying clone and renal function.
FundingThe present study was supported in part by grants from the Instituto de Salud Carlos III co-funded by the Fondo Europeo de Desarrollo Regional – FEDER (RD16/0009/0006; grant ICI14/00016 and grant PI17/02043) from the Spanish Ministry of Economy and Competitiveness. These institutions had no part in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Authors’ contributionsAll the authors contributed to the manuscript. J.A.-T., MD, M.-E., V.L., M.L., G.M.-R., P.R.-E., D.H., wrote the manuscript, contributed to discussion and reviewed/edited the manuscript. All authors have read and approved the final version.
Availability of data and materialData sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Conflict of interestThe authors declare no conflicts of interest.
We thank Maria Repice and Ian Johnstone for linguistic assistance in the preparation of the text.



