







El hiperparatiroidismo secundario es uno de los componentes integrales de las alteraciones del metabolismo óseo-mineral en la enfermedad renal crónica (ERC) o complejo chronic kidney disease-mineral bone disorder. Se ha demostrado que en el desarrollo y progresión del hiperparatiroidismo secundario intervienen muchos factores, estrechamente interrelacionados, pero la presencia e importancia de la hiporrespuesta (o resistencia) a la acción de la hormona paratiroidea (PTH) es poco comprendida. En esta revisión analizaremos sus antecedentes, factores que intervienen, así como alguno de los mecanismos moleculares que podrían explicarla. La presencia de resistencia a la acción biológica de la PTH no es única en la ERC, ya que también se presenta para otras hormonas, habiéndose incluso usado el término de «uremia como una enfermedad de receptores». Esta hiporrespuesta a la PTH tiene importantes implicaciones clínicas, dado que no solo permite explicar parte de la patogenia progresiva de la hipersecreción de PTH e hiperplasia paratiroidea, sino también la creciente prevalencia de enfermedad ósea adinámica en la población con ERC. De este modo, subrayamos la importancia de controlar, sin normalizar completamente, los niveles de PTH en los distintos estadios de ERC, dado que un cierto incremento de sus niveles supone inicialmente una adaptación clínica. Futuros estudios a nivel molecular sobre la uremia, o la reciente descripción del efecto directo del fosfato sobre la actividad del receptor sensor de calcio como sensor de fosfato, podrían resultar valiosos incluso más allá de explicar la hiporrespuesta a la PTH en la ERC.
Secondary hyperparathyroidism (SHPT) is an integral component of the chronic kidney disease–mineral and bone disorder (CKD–MBD). Many factors have been associated with the development and progression of SHPT but the presence of skeletal or calcemic resistance to the action of PTH in CKD has often gone unnoticed. The term hyporesponsiveness to PTH is currently preferred and, in this chapter, we will not only review the scientific timeline but also some of the molecular mechanisms behind. Moreover, the presence of resistance to the biological action of PTH is not unique in CKD since resistance to other hormones has also been described (“uremia as a receptor disease”). This hyporesponsiveness carries out important clinical implications since it explains, at least partially, not only the progressive nature of the pathogenesis of CKD-related PTH hypersecretion and parathyroid hyperplasia but also the increasing prevalence of adynamic bone disease in the CKD population. Therefore, we underline the importance of PTH control in all CKD stages, but not aiming to completely normalize PTH levels since a certain degree of SHPT may represent an adaptive clinical response. Future studies at the molecular level, i.e. on uremia, or the recent description of the calcium-sensing receptor as a phosphate sensor, may become of great value beyond their significance to explain just the hyporesponsiveness to PTH in CKD.
La enfermedad renal crónica (ERC) es un problema de salud global importante con un extremado riesgo cardiovascular y de mortalidad1,2, en parte relacionado con las alteraciones del metabolismo óseo-mineral (chronic kidney disease-mineral bone disorder [CKD-MBD]). El síndrome/complejo CKD-MBD incluye alteraciones bioquímicas, óseas y/o calcificaciones cardiovasculares, estrechamente interrelacionadas3, mientras que el término «osteodistrofia renal» se considera actualmente solo una medida del componente óseo cuantificable por histomorfometría3. Actualmente también hemos descrito el hueso como un órganoendocrino, productor de hormonas con acciones sistémicas (por ejemplo, fibroblast growth factor 23 [FGF23], esclerostina y osteocalcina), potencialmente responsables de complicaciones metabólicas y cardiovasculares que presentan los pacientes con ERC4.
El aumento progresivo de los niveles de hormona paratiroidea (PTH) (hiperparatiroidismo secundario [HPS]) es uno de los componentes del complejo CKD-MBD y, dejado a su libre evolución, condicionará el empeoramiento de otras alteraciones bioquímicas (p. ej. hipercalcemia y/o hiperfosfatemia), estructura ósea (enfermedad ósea de alto recambio) y se asociará a trastornos cardiovasculares, entre otros efectos5,6. Clásicamente, la PTH se ha considerado como una «toxina urémica» cuyos efectos no se limitan solo al hueso7,8. De hecho, la PTH produce un aumento de calcio (Ca) intracelular en diferentes tipos de células y estimula la producción de FGF23 (con efectos sistémicos claramente demostrados). La mejoría de algunos de estos efectos deletéreos tras paratiroidectomía (PTX) respaldaría la presencia de un vínculo causal, al menos indirecto, con la PTH8,9. Estos potenciales efectos tóxicos de la PTH podrían también explicar la asociación del HPS con la progresión de la ERC, la enfermedad y mortalidad cardiovascular ateromatosa y no ateromatosa, así como su asociación con mortalidad por todas las causas10–12. Es interesante observar que más allá de la ERC algunas cohortes en pacientes cardiológicos han confirmado la asociación independiente entre niveles altos de PTH, eventos cardiovasculares y mortalidad (incluida la muerte súbita)13.
El HPS es una complicación clásica, progresiva, muy frecuente y potencialmente grave de la ERC, pero por otro lado los niveles de PTH muy bajos o relativamente bajos también se han asociado a una alta morbimortalidad10,14,15. El vínculo entre una PTH relativamente disminuida y la presencia de un bajo remodelado óseo (siendo actualmente su manifestación más frecuente la enfermedad ósea adinámica [EOA]) es bien conocido16, y la reciente asociación a morbimortalidad pudiera estar relacionada con un aumento de fracturas-microfracturas óseas y/o la aparición o progresión de las calcificaciones cardiovasculares15,17–23. Por lo tanto, no es sorprendente que la aceptada asociación entre mortalidad y PTH sea compleja y solo evidente en ambos extremos (niveles elevados y bajos) de PTH3,10. Estudios de cohortes muestran que solo los niveles extremos de PTH pueden predecir, con una sensibilidad/especificidad aceptables, el recambio óseo subyacente (EOA [bajo recambio]/osteítis fibrosa [alto recambio])24. La combinación de los niveles de PTH con los de fosfatasa alcalina (especialmente su fracción ósea) podría mejorar esta capacidad de predicción, pero aún está muy lejos del «patrón oro» (biopsia ósea)25–28.
Fisiopatología del hiperparatiroidismo secundarioLa pérdida progresiva de la función renal conduce a una disminución de la expresión del receptor de la PTH (PTHR1) y α-klotho en el riñón, del PTHR1 en el hueso, elevación del FGF23, retención de P29–31 y una reducción de la síntesis/aumento del catabolismo del calcitriol (1,25-dihidroxivitamina D; vitamina D activa), entre muchos otros trastornos metabólicos32,33.Todos estos factores, ampliamente revisados en otros artículos32,34,35, involucran a varios mecanismos interrelacionados, incluyendo la disminución de la expresión de receptores en células paratiroideas (receptor sensor de Ca [RSCa], receptor de vitamina D [RVD], receptor del FGF [FGFR]-klotho), y que son responsables de la inhibición de la síntesis, secreción de PTH y su capacidad de proliferación (policlonal inicialmente, potencialmente monoclonal si persisten estímulos pro-proliferativos). Se sabe que la hiperplasia paratiroidea se desarrolla progresivamente desde los estadios iniciales de la ERC7,32,36 y en la ERC avanzada, a pesar del aumento del FGF23 y PTH, la excreción renal de P es insuficiente y se produce un aumento de P extracelular que directa e indirectamente estimula la secreción de PTH37–40.
El Ca extracelular es el regulador más importante de la función de la glándula paratiroidea8,36. Para corregir una potencial hipocalcemia es inicialmente necesaria la liberación rápida (secreción) de la PTH almacenada en la célula paratiroidea, seguido de un aumento de la síntesis de nueva PTH para, finalmente, necesitar del incremento del número de células paratiroideas funcionantes si el estímulo hipocalcémico permanece en el tiempo. De hecho, los mecanismos mencionados anteriormente (p. ej. retención de P y disminución de calcitriol) tendrían en común la capacidad de producir hipocalcemia secundaria (p. ej. disminución especular de Ca plasmático por hiperfosfatemia y disminución de absorción intestinal) e inducir el HPS. En la célula paratiroidea la hipocalcemia mantenida reduce la expresión de receptores inhibitorios de su actividad (RSCa, RVD, FGFR1-klotho), induciendo un aumento de la síntesis y secreción de PTH, además de proliferación celular34,35,41,42. En esta revisión, más allá de analizar las complejas interacciones fisiopatológicas de los distintos factores que conducen al HPS, nos centraremos en la importancia de la hiporrespuesta (resistencia calcémica o esquelética) a la acción de la PTH en la patogénesis del HPS y hasta qué punto puede contribuir al desarrollo de EOA.
Resistencia o hiporrespuesta a la acción de la hormona paratiroideaLa resistencia a la acción de la PTH en la ERC (también conocida como resistencia esquelética, resistencia ósea, disminución de la respuesta calcémica de la PTH o, simplemente, resistencia a la PTH) es un concepto antiguo43, actualmente renombrado como «hiporrespuesta» a la PTH44. De hecho, las respuestas tanto óseas como renales a la acción de la PTH están progresivamente deterioradas en la ERC44 y el término «hiporrespuesta» puede ser más apropiado, dado que la respuesta a la PTH está mitigada pero no completamente ausente44 (tabla 1).
Terminología
| Hiporrespuesta a la PTH | Término genérico actual que expresa una disminución de la respuesta biológica normal a la acción de la PTH propia de la ERC. Puede ponerse de manifiesto como disminución de la respuesta calcémica a una infusión de PTH (en modelos experimentales) o como un hueso con disminución del remodelado en relación con la cantidad de PTH presente (p. ej. con biopsias óseas). Para que haya una hiporrespuesta debe haber una disminución del número o una disfunción de las células óseas |
| Disminución de la respuesta calcémica (esquelética) a la acción de la PTH o resistencia a la PTH | Demostración experimental de la existencia de una hiporrespuesta a la PTH. La infusión de una cantidad fija de PTH en el animal experimental produce un esperado aumento del calcio plasmático, pero de magnitud diferente con la modificación de distintos factores (grado de función renal, contenido de fósforo en la dieta, etc.). Los animales experimentales reciben una dieta sin calcio durante la infusión, por lo que el aumento de calcio plasmático se considera equivalente a su acción esquelética |
ERC: enfermedad renal crónica; PTH: hormona paratiroidea.
Fue descrita por primera vez por J.M. Evanson en 196643 en 12 pacientes hipocalcémicos con ERC43. Posteriormente, Massry et al.45 observaron que la respuesta calcémica a un extracto de paratiroides estaba marcadamente disminuida en perros paratiroidectomizados después de la inducción de uremia, y que la respuesta calcémica a la PTH fue menor en pacientes con ERC moderada y avanzada (incluidos pacientes en hemodiálisis y trasplante renal)46. Llach et al. describieron una disminución de la respuesta calcémica a la PTH endógena47,48. Dichas observaciones indicaron que esta respuesta calcémica alterada a la PTH aparece en fases precoces de la ERC, siendo una consecuencia directa el requerimiento continuo de una mayor concentración de PTH circulante para mantener la homeostasis del Ca. Desde el antiguo estudio de Albright et al.49 y la hipótesis del trade-off de Bricker y Slatopolsky30,50, ya se había planteado que la retención de P y la disminución recíproca de Ca inducirían hiperplasia paratiroidea y osteítis fibrosa ósea. La presencia de resistencia esquelética a la PTH en la ERC proporcionaría un mecanismo adicional amplificador, generalmente olvidado, sobre la patogénesis de la hipocalcemia y HPS progresivo en la ERC32,51,52. La aparición de resistencia esquelética a la acción de la PTH al inicio del curso de la ERC contribuiría, junto a otros factores (hiperfosfatemia, déficit de vitamina D, etc.), al desarrollo de hipocalcemia, que a su vez estimularía las glándulas paratiroideas, aumentando la secreción de PTH y/o induciendo hiperplasia glandular. En el otro extremo la aparición tardía de hipocalcemia, solo detectable en etapas avanzadas de ERC, representaría claramente la etapa final de la incapacidad de la PTH para restablecer los niveles de Ca a la normalidad y así reflejar la máxima expresión clínica de hiporrespuesta a la PTH.
Factores vinculados a la hiporrespuesta a la hormona paratiroidea en la enfermedad renal crónicaSon múltiples los factores que han sido asociados de un modo u otro a la hiporrespuesta a la PTH en la ERC, ya sean relacionados con alteraciones del metabolismo mineral en el contexto de la ERC o el efecto indefinido de la acumulación de productos (conocidos o no) en fases más avanzadas («uremia»), así como diversas circunstancias demográficas (raza, edad y sexo) o patológicas (p. ej. diabetes), entre muchos otros (fig. 1).
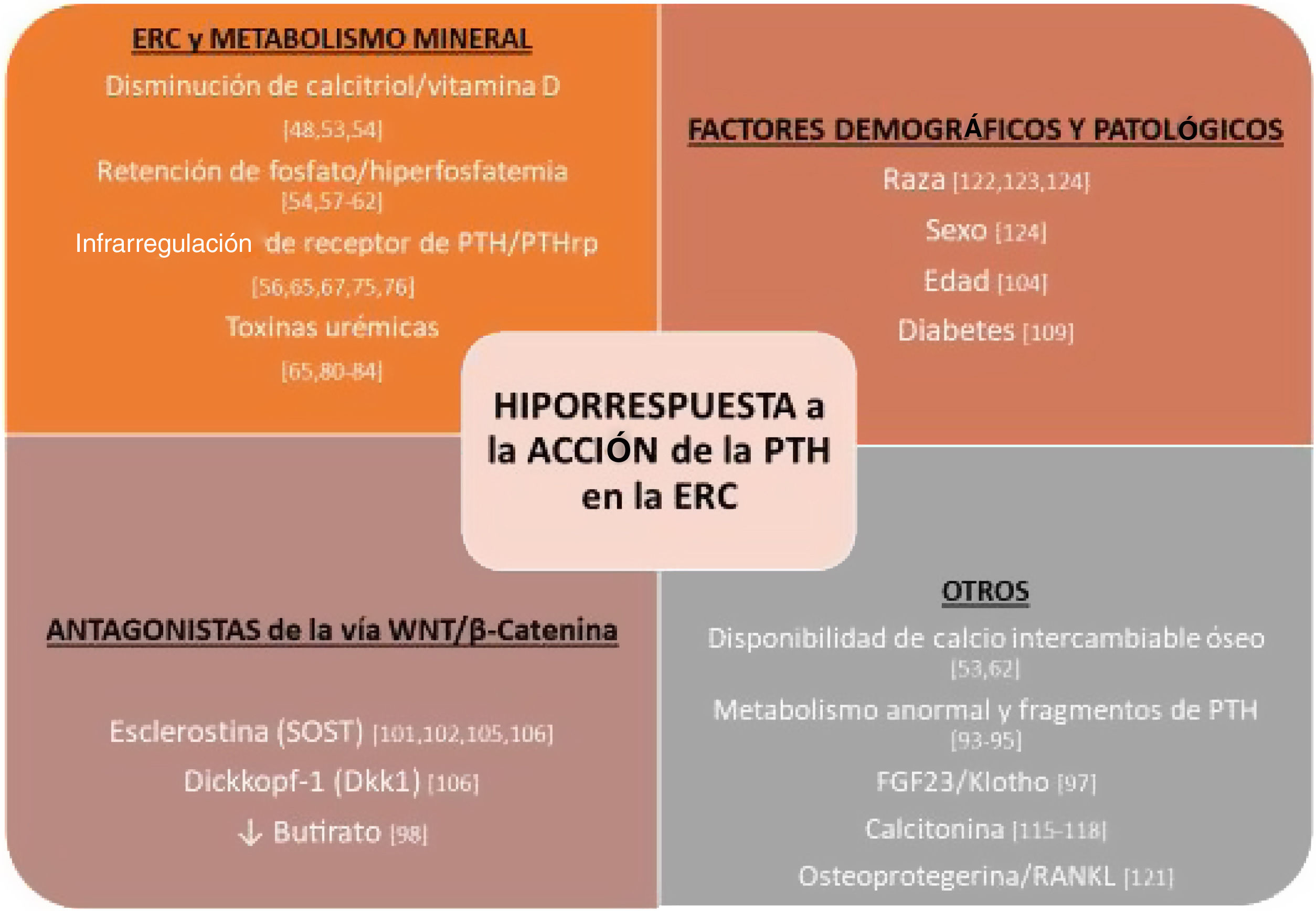
Resumen de factores implicados en la hiporrespuesta a la acción de la hormona paratiroidea (PTH) en la enfermedad renal crónica (ERC).
FGF-23: acrónimo inglés del fibroblast growth factor23; PTHrp: péptido relacionado con la PTH; RANKL: acrónimo inglés del receptor activator for nuclear factor κB ligand.
Desde el primer trabajo en este campo se consideró que la vitamina D era necesaria para la acción adecuada de la PTH en el hueso43.Posteriormente, se demostró que la administración de calcitriol restableció, al menos parcialmente, la respuesta calcémica a la PTH en animales experimentales53,54, lo que llevó a la conclusión de que estaría relacionada con la disminución de los niveles de calcitriol de los estadios iniciales de la ERC. Además, en pacientes con ERC precoz la administración diaria de calcitriol durante 6 semanas mejoró la respuesta calcémica a la PTH48. Finalmente, estudios experimentales también mostraron una mejoría después de la administración de calcitriol junto con 24,25(OH)2D355. Por ello, aunque todavía faltaría una explicación adecuada sobre el mecanismo de acción, parecería que la vitamina D podría mejorar la acción de la PTH en el hueso. Sin embargo, otros investigadores no confirmaron este efecto53,56.
Retención del fosfatoDesde los trabajos clásicos de Somerville y Kaye, así como otros con modelos experimentales diferentes54,57, es bien sabido que la retención de P disminuye muy significativamente la respuesta calcémica a la PTH en la ERC. Dado que el P y el calcitriol también están estrechamente relacionados entre sí (el P inhibe la síntesis de calcitriol), no se pudo excluir que al menos algunas de estas observaciones pudieran estar mediadas indirectamente por la inhibición del calcitriol. También se demostró que las ratas con ERC alimentadas con una dieta baja en P presentaron una mejoría de la respuesta calcémica durante una infusión constante estandarizada de PTH 1-34, pero solo aquellas con ERC moderada tuvieron un incremento significativo de los niveles de calcitriol. De este modo, la restricción de P mejoró la respuesta calcémica a la PTH pero, en la ERC avanzada, la respuesta calcémica mejoró con la restricción de P independientemente del calcitriol. De hecho, en estudios posteriores demostramos que el efecto negativo de la retención de P en la respuesta calcémica a la PTH puede ser muy superior al efecto de la deficiencia de calcitriol58,59. La mejoría de la respuesta calcémica a una infusión de PTH después de la restricción de P también se ha demostrado en pacientes con ERC leve60.
Curiosamente, en ratas con función renal normal que recibieron una dieta alta en P, la respuesta calcémica a la PTH se redujo en ausencia de cambios en los niveles séricos de P, indicando que el contenido de P en la dieta misma, directa o indirectamente, fue responsable de la reducción de la respuesta54,61. Este es un tema a tener en cuenta actualmente, dada la falta de una recomendación clara sobre si la restricción de P en la dieta debe aplicarse en estadios 2-3 de la ERC. El mecanismo intrínseco que conduce a una disminución en la respuesta calcémica a la PTH inducida por P no se conoce completamente, pero es probable que, además del efecto negativo del P sobre los niveles de calcitriol, la concentración de P ambiental en el hueso podría afectar la cantidad de Ca intercambiable del hueso que pudiera ser movilizado por la PTH53,62.
Todos los experimentos mencionados se realizaron antes del descubrimiento del FGF23, Dickkopf-1 (Dkk1) o la esclerostina y, hasta la fecha, no se ha excluido la posibilidad de que niveles elevados de estas moléculas pudieran reducir el efecto de la PTH sobre el hueso. Podría ser que FGF23, directa o indirectamente a través de la supresión del calcitriol, o la activación del Dkk1 o la esclerostina (inhibidores de la vía del Wnt/ß-catenina) pudieran interferir con el flujo de Ca mediado por la PTH en el hueso44.También se han descrito recientemente múltiples puntos de unión, no solo para Ca2+, sino también para PO43− en el RSCa que podría modularse por la concentración de P (sensor de P)63,64. Mientras que el Ca2+ activa el RSCa, el PO43− refuerza su conformación inactiva, contribuyendo así a una mayor secreción de PTH63. De la misma forma que el P interfiere con la activación del RSCa en la célula paratiroidea, podría también interferir con el RSCa de los osteoblastos y osteoclastos, pero esto no ha sido investigado. En definitiva, la interacción del P sobre el RSCa óseo podría interferir con la respuesta calcémica a la PTH en la ERC.
Aunque tanto la restricción de P como la administración de calcitriol mejoran la respuesta calcémica a la PTH, no parece que la recuperen completamente, ya sea de modo aislado o en combinación. Por el contrario, en animales con PTX o tiroparatiroidectomizados54,56,65, la eliminación de la PTH circulante corrige sorprendentemente la respuesta calcémica a la PTH, aun a pesar de la presencia de hiperfosfatemia y niveles bajos de calcitriol.
Infrarregulación de los receptores de la hormona paratiroideaSe ha descrito previamente en animales urémicos que la elevación de la PTH endógena podría desensibilizar el esqueleto a la administración de PTH exógena a través de la infrarregulación de receptores óseos (como un mecanismo de defensa)56,66. El potencial papel de la infrarregulación de receptores óseos se postuló especialmente tras la «sorprendente» restauración de la respuesta calcémica a la PTH tras PTX mencionada. Asimismo, en huesos de perros urémicos, con insuficiencia renal aguda o crónica, se demostró la reducción de la liberación de AMP cíclico tras la administración de PTH, y que esta se restauraba tras tiroparatiroidectomía67,68.
Aunque a nivel experimental la PTX corrige la respuesta calcémica a la PTH, nosotros observamos que mantener los niveles de PTH en el rango normal en ratas con ERC que recibían una dieta baja en P no la restauró65.Además, los animales urémicos con niveles normales de PTH, obtenidos tras PTX parcial, mostraron todavía una disminución del 50% en la respuesta calcémica a la PTH en comparación con ratas normales65. Esto es consistente con estudios clínicos que habían mostrado que la PTX subtotal casi normalizó los niveles de PTH, pero no mejoró la respuesta calcémica a la PTH46. Además, la PTX mejoró la respuesta calcémica a la PTH no solo en animales con ERC, sino también en los animales usados como control65. Por todo ello, la infrarregulación de los receptores de PTH tras una excesiva exposición no parecería ser la única explicación a la restauración de la respuesta calcémica tras PTX. Además de la explicación clásica de que la restauración de esta tras PTX se debería a un fenómeno de hipersensibilización, al igual que se describe en diferentes sistemas hormonales, es posible que la PTX pudiera también afectar a la acumulación o distribución del Ca disponible en el hueso, como ha sido descrito en otros estudios69.
Todos estos hallazgos no excluyen la infrarregulación de los receptores de PTH como una causa potencial de disminución de la respuesta calcémica a la PTH70. La clonación del gen del receptor tipo 1 de la PTH (PTHR1), un receptor común para PTH y péptido relacionado con PTH (PTHrP)71,72, ha permitido demostrar que el receptor 1 de la PTH (PTH1R) no solo está ampliamente distribuido en tejidos73, sino que también está infrarregulado en riñones urémicos y hueso74–77. Los datos humanos son menos consistentes, ya que algunos investigadores demostraron una disminución de su expresión, mientras que otros describieron un aumento. Distintas metodologías y características de las poblaciones estudiadas podrían explicar esta aparente discrepancia.
Por otra parte, el hallazgo de infrarregulación del ARN mensajero (ARNm) del PTH1R en tejido óseo y no en el hígado o el corazón sugirió que la expresión de PTH1R se regula de una manera específica en la célula, independientemente del estado urémico78. Sin embargo, es probable que otros factores, además del aumento de los niveles de PTH, pudieran disminuir la actividad del receptor75,78. De este modo, se ha demostrado que ni el aumento de la PTH y del P ni la disminución del Ca plasmático sería importante en la infrarregulación del PTH1R renal durante la ERC, así como que también parecería poco probable que fuera secundario a un aumento del PTHrP renal75. Sin embargo, otros autores observaron una mayor expresión de ARNm de PTH1R después de PTX, pero sin controles76.
Aunque los mecanismos responsables de la supuesta desensibilización o infrarregulación del PTH1R en la ERC siguen estando muy mal definidos, algunos estudios han implicado varios factores urémicos y fragmentos de la PTH C-terminal (ver más adelante) en este fenómeno. La información disponible sobre la regulación del PTH1R es muy limitada e incluso contradictoria, y se encuentra más allá del alcance de esta revisión79.
Toxinas urémicasEs importante destacar que, en ratas con ERC, observamos una disminución significativa en la respuesta calcémica a una infusión de PTH a pesar de la presencia de niveles séricos normales de Ca, P, PTH e incluso calcitriol (fig. 2). Esta respuesta calcémica refleja directamente la hiporrespuesta esquelética a la acción de la PTH, dado que los animales reciben durante la infusión una dieta sin calcio. Este hallazgo nos permitió inferir que factores intrínsecos a la uremia per se (conocidos o no) disminuían la respuesta calcémica a la PTH65. Estos datos se confirmaron posteriormente en un modelo diferente cuando se observó que el mantenimiento de niveles normales de PTH en ratas urémicas con PTX, a las que se administró una infusión constante de PTH, tampoco corregía la respuesta calcémica80.
![Hiporrespuesta a la acción de la hormona paratiroidea (PTH). Se muestra la respuesta calcémica (calcio [Ca] plasmático en mg/dl) a la infusión durante 48 horas de una cantidad constante de PTH en ratas con diferentes grados de función renal (normal, insuficiencia renal moderada e insuficiencia renal avanzada) y diferente contenido de fósforo en la dieta previa (AP: dieta alta en fósforo [1,2%]; BP: dieta baja en fósforo [0,3%]; MP: dieta moderada en fósforo [0,6%]. Durante la infusión las ratas no reciben calcio en la dieta, por lo que el incremento de calcio se debe a la respuesta esquelética a la infusión de PTH. Se aprecia cómo la respuesta calcémica es una situación dinámica cuya magnitud, entre otros factores, depende del grado de función renal (menor respuesta calcémica a mayor deterioro renal) y la cantidad de fósforo en la dieta (menor respuesta calcémica a mayor contenido en fósforo en la dieta [y fósforo sérico, no representado]) (adaptado de Bover et al.58 y Bover et al.59). El término respuesta calcémica o resistencia esquelética a la PTH ha sido reemplazado por el de hiporrespuesta a la acción de la PTH44.](https://static.elsevier.es/multimedia/02116995/0000004100000005/v1_202109170826/S0211699521000709/v1_202109170826/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92lL8PRnKGMnsiVXV6EVJ5QRkjJZIKj2umwUyY+cL6K8EPtBNKuUo2iJd6fqhiK874gsPvaYt8HKeK98oH5m8LEiJSPhLeB24KDpZYGGcEvEfAExiQv0HltDbSwmKw82X5YIJ4Tr3/g6pNRB7oRJKHj/cwW3AW3piQ3r0DRDo+snp+T4YLivOMz3/OSTIpPTRugw/DegxvltNndz4ETdWoVwyX1RnyyyxifBDS9d+V3Ms2PRQS+54WybW5N0grUVEjWaeXgNu5tp0Yy6WH/jH4o=)
Hiporrespuesta a la acción de la hormona paratiroidea (PTH). Se muestra la respuesta calcémica (calcio [Ca] plasmático en mg/dl) a la infusión durante 48 horas de una cantidad constante de PTH en ratas con diferentes grados de función renal (normal, insuficiencia renal moderada e insuficiencia renal avanzada) y diferente contenido de fósforo en la dieta previa (AP: dieta alta en fósforo [1,2%]; BP: dieta baja en fósforo [0,3%]; MP: dieta moderada en fósforo [0,6%]. Durante la infusión las ratas no reciben calcio en la dieta, por lo que el incremento de calcio se debe a la respuesta esquelética a la infusión de PTH. Se aprecia cómo la respuesta calcémica es una situación dinámica cuya magnitud, entre otros factores, depende del grado de función renal (menor respuesta calcémica a mayor deterioro renal) y la cantidad de fósforo en la dieta (menor respuesta calcémica a mayor contenido en fósforo en la dieta [y fósforo sérico, no representado]) (adaptado de Bover et al.58 y Bover et al.59). El término respuesta calcémica o resistencia esquelética a la PTH ha sido reemplazado por el de hiporrespuesta a la acción de la PTH44.
Ya se había postulado previamente que la presencia de factores urémicos desconocidos, más allá del P, podrían ser responsables de la disminución de la respuesta calcémica a la PTH en la ERC46. Posteriormente, Wills y Jenkins también demostraron que el suero de pacientes urémicos inhibió la resorción ósea inducida por la PTH en un modelo in vitro, mientras que el suero obtenido después de la diálisis no lo hizo81. Se han descrito también inhibidores de bajo peso molecular de la mitogénesis de osteoblastos en plasma urémico82, y estudios experimentales posteriores apuntaron hacia diferentes toxinas urémicas83 que desencadenan estrés oxidativo, como el indoxil sulfato (IS) y p-cresil sulfato84,85 y/o lipoproteínas de baja densidad oxidadas proinflamatorias86. El aumento del estrés oxidativo y la inflamación de bajo grado son comunes en la ERC y, por lo tanto, pueden tener carácter causal, pero podrían afectar vías comunes de señalización (p. ej. P, FGF23/klotho o la deficiencia de calcitriol también se asocian con estrés oxidativo e inflamación)44,87. Estudios in vitro e in vivo han demostrado la posible asociación de toxinas urémicas (i. e. IS) a situaciones de bajo recambio óseo88,89. Nii-Kono et al.84 demostraron además que el IS induciría un estado de resistencia a la PTH, reduciendo la expresión de AMPc y del PTH1R inducido por la PTH, disminuyendo la viabilidad de los osteoblastos en cultivo. Estos autores también demostraron que la producción de radicales libres en los osteoblastos aumentaba en relación con la concentración de IS añadida84. Además, sus resultados sugirieron que los osteoblastos captan el IS a través de un transportador de aniones, lo que aumentaría el estrés oxidativo, alteraría la función de los osteoblastos y regularía la expresión de PTH1R84. Sin embargo, los resultados de correlacionar los niveles de IS con la histomorfometría ósea de pacientes con ERC resultan contradictorios90. Por otra parte, se ha descrito que algunas toxinas urémicas (por ejemplo el ácido úrico) podrían estimular indirectamente la secreción de PTH al disminuir la síntesis de calcitriol y unirse a los elementos de respuesta de la vitamina D en el ADN91, induciendo resistencia a la acción de otra hormona como el calcitriol. En una muy reciente revisión92 se amplía el papel del síndrome urémico sobre distintas vías de señalización, describiéndose relaciones interorgánicas aberrantes y mostrando cómo distintas toxinas urémicas pueden interferir vías metabólicas, múltiples moléculas y transportadores92.
Metabolismo anormal de los fragmentos de la hormona paratoriodea en la enfermedad renal crónicaEl aumento de secreción de PTH es el principal responsable de los niveles plasmáticos elevados de PTH en la ERC. Sin embargo, los riñones desempeñan un papel importante en la degradación de la misma y, en los pacientes con ERC, se reduce el aclaramiento de la PTH, al igual que el de otras hormonas peptídicas (p. ej. insulina).
Cómo la ERC afecta la secreción, pulsatilidad, metabolismo y degradación de la PTH (sus fragmentos amino- y/o carboxi-terminales, también presentes en otras moléculas [i. e. FGF23]) y sus vías de señalización están ampliamente tratados en otras revisiones más específicas32,44, así como la forma en que estos condicionan la dificultad para definir niveles de PTH óptimos para los distintos estadios de la ERC3.
Finalmente, es importante resaltar que los fragmentos carboxiterminales de la PTH no solo no son biológicamente inactivos sino que, al ocupar el PTHR1, tienen efectos antagonistas a la PTH en el riñón y el hueso93. En pacientes con ERC la presencia de niveles circulantes elevados de estos fragmentos de PTH detectados por el ensayo habitual de PTH «intacta», pero con efectos antagonistas, explicaría también la necesidad de niveles más altos de PTH para prevenir la EOA, lo que supuso en su momento un mecanismo novedoso para explicar la «resistencia» esquelética a la acción de la PTH en la uremia93.
Además del PTH1R «clásico», actualmente se acepta que un PTHR carboxiterminal (PTH4R o PTHR-C) podría mediar algunas acciones dispares de la PTH94,95. También se han descrito otros receptores de PTH (PTH2R y PTH3R), pero sus efectos en humanos y en ERC solo son conocidos parcialmente96.
Cascada de señalización de la hormona paratiroidea, factores locales y sistémicosComo hemos mencionado con anterioridad, la PTH ejerce su acción al unirse al PTHR1, expresado fundamentalmente en osteoblastos y osteocitos además del riñón44. Dicha unión desencadena una cascada de transducción de señales por diversas vías (p. ej. proteín-cinasas A y C, Wnt/ß-catenina, etc.) que promoverán diferentes respuestas celulares. Es posible que los efectos resultantes de esta señalización subsecuente de la PTH, ’downstream signaling’, se vean contrarrestados por otras señales paracrinas o endocrinas inhibitorias competidoras inducidas por el FGF23, klotho, antagonistas de la vía del Wnt/ß-catenina (como la esclerostina), calcitonina, osteoprotegerina, proteínas morfogenéticas óseas, entre otras moléculas, sumados a otros efectos locales (inflamación, citoquinas, estrés oxidativo, alteraciones en el equilibrio ácido-base o las concentraciones de Ca, P, magnesio, flúor o aluminio)33,44. De hecho, un estudio reciente demostró cómo la proteína recombinante humana α-klotho interaccionaba con el PTH1R inhibiendo la unión de la PTH humana a las células tubulares renales97. A su vez, inhibía la expresión de 1α-hidroxilasa inducida por PTH a este nivel. Estos resultados sugerían que α-klotho podría mediar en la inhibición de la síntesis de calcitriol inducida por FGF23 al bloquear la señalización de PTH97. Otro ejemplo más complejo y reciente sería el papel del butirato procedente de la microbiota intestinal. Es sabido que la depleción de la microbiota disminuye los niveles de butirato, metabolito responsable de la comunicación hueso-intestino98. Así, se ha demostrado que el restablecimiento de niveles fisiológicos de butirato restauró la acción anabólica de la PTH a través de la activación de la vía osteogénica Wnt/ß-catenina dependiente de células T98.
En este sentido, creemos que es muy importante destacar el potencial papel de los antagonistas de la vía del Wnt/ß-catenina al ser la PTH un agonista de esta vía99. La esclerostina, producida principalmente por los osteocitos (al igual que el FGF23 o la osteocalcina), fue considerada originalmente como un antagonista no clásico de las proteínas morfogenéticas óseas100. Actualmente la esclerostina ha sido identificada como un inhibidor soluble de esta vía de señalización al fijarse a los receptores LRP5/6101,102, produciendo una disminución en la formación de hueso al inhibir la osteoblastogénesis. Además, parece desempeñar un papel como mediadora de otros factores sistémicos y locales tales como calcitriol, PTH, glucocorticoides y el factor de necrosis tumoral-α (TNFα)103. Pese a todo, se sigue debatiendo hasta qué punto los niveles circulantes de esclerostina reflejan su expresión ósea o cómo afectan otras vías de señalización local104. De hecho, el aumento de la expresión de esclerostina se ha visto en pacientes con ERC incipiente, pese a tener niveles normales de PTH105, y este aumento persiste en pacientes en diálisis, aunque en menor medida, a pesar de tener niveles elevados de PTH106. Es posible que, en un futuro próximo, la relación (cociente) entre el agonismo de la vía Wnt/ß-catenina (p. ej. por PTH) y el antagonismo (p. ej. por esclerostina, Dkk1 u otras moléculas asociadas) permita avanzar en el conocimiento de las causas de la hiporrespuesta a la PTH en la ERC y/o facilite el diagnóstico no invasivo del recambio óseo.
Los niveles circulantes de esclerostina aumentan con la edad y con la disminución de la función renal103,107,108, estando incrementados también en pacientes diabéticos, independientemente de la edad o del sexo109. La esclerostina y las moléculas relacionadas con ella, como la Dkk1 o la frizzled 4 sérica, se han postulado como posibles mediadores en el desarrollo de EOA33,85 y/o en la resistencia esquelética a la acción de la PTH33,44,85. De este modo se ha propuesto que la inhibición temprana de la vía del Wnt/ß-catenina de los osteocitos por el aumento de estos factores en la ERC podría ser el estadio inicial de la osteodistrofia renal, y podría explicar el aumento de prevalencia de la EOA90,110–112. Así, se ha hipotetizado que en estadios iniciales de la ERC podría prevalecer la enfermedad ósea de bajo recambio, siendo la EOA la forma predominante33,85. Con la progresión de la ERC a estadios más avanzados, el aumento de niveles circulantes de PTH y el incremento paulatino de activación del PTH1R eventualmente superarían la resistencia esquelética a la PTH y, sin tratamiento, generarían la clásica osteítis fibrosa8,33. No se conoce si FGF23 y/o α-klotho ejercerían un papel directo en esta transición teórica de bajo a alto remodelado óseo o participarían únicamente de forma indirecta a través de la regulación de la secreción de PTH33. De todos modos, la disfunción de los osteocitos se ha descrito ya alterada en el curso inicial de la ERC33.Curiosamente, el uso de anticuerpos anti-esclerostina (romosozumab) en modelos de ratas con ERC con osteodistrofia renal progresiva parecía incrementar el volumen total de hueso trabecular y la superficie de mineralización trabecular solo en animales con niveles bajos de PTH113. Del mismo modo, las propiedades óseas (volumen óseo, geometría cortical y propiedades bioquímicas) mejoraron solamente cuando los niveles de PTH se mantenían bajos113. Quedaría por aclarar si los niveles elevados de esclerostina son causa o consecuencia de la hiporrespuesta a la PTH en la ERC114.
Desde el punto de vista molecular cabe mencionar también que han sido analizados el papel potencial de la producción endógena de calcitonina (producida por las células C de la glándula tiroidea) en la patogenia del HPS y su papel sobre la respuesta a la PTH en la ERC115–120, así como el de la elevación de los niveles de osteoprotegerina, que podrían favorecer la resistencia esquelética a la PTH a través de la supresión de la osteoclastogénesis121.También es necesario recordar, como hemos mencionado con anterioridad, que distintas toxinas urémicas per se se han asociado a hiporrespuesta a la PTH en la ERC quizás interfiriendo en distintas vías metabólicas, modulando la actividad de las quinasas (fosforilación) y/o, como se recalca en este apartado, uniéndose a segundos mensajeros, afectando vías intracelulares que pueden afectar la transcripción, el tráfico de proteínas e interacciones moleculares91,92.
Finalmente, se debe también tener en cuenta que otros factores como la raza, las diferencias de género122–124, la edad avanzada o el aumento de prevalencia de diabetes (conocida causa de EOA) en la población con ERC, además del propio recambio óseo o el control exhaustivo de la PTH (erróneamente normalizando los niveles de PTH en pacientes con ERC que aún no están en diálisis) parecen influir en la evaluación de dicha hiporrespuesta a la PTH125.
Implicaciones clínicas de la hiporrespuesta a la hormona paratiroideaComo hemos visto, la resistencia esquelética (calcémica) a la PTH fue inicialmente sugerida como un mecanismo implicado en la patogenia del HPS en la ERC. Serían necesarios niveles de PTH progresivamente más elevados durante la evolución de la ERC para intentar normalizar el calcio plasmático, complementando así la trade-off hypothesis30,50,51. El interés sobre el concepto de resistencia a la PTH ha resurgido con la supresión potencialmente excesiva de la PTH con distintas terapias (p. ej. quelantes de P basados en Ca, vitamina D, calcimiméticos y PTX) y al aumento observado de la prevalencia de EOA con sus riesgos asociados, previamente mencionados15,20–22,44.También se ha descrito recientemente que los mismos niveles de PTH conseguidos con distintas terapias (p. ej. calcimiméticos vs. vitamina D) podrían tener distinto significado clínico al tener dichos fármacos acciones sobre el hueso independientes de su efecto sobre la PTH126,127.
Ya dijimos que la resistencia esquelética a la PTH es conocida actualmente como «hiporrespuesta» a la PTH o «hipoparatiroidismo relativo», dado que la respuesta a la PTH está mitigada pero no completamente ausente. London et al., analizando la presencia de una intercomunicación entre hueso y vasos128, ya habían mostrado una asociación inversa entre calcificación vascular y niveles disminuidos de PTH, un descenso del número de osteoclastos, menor superficie osteoblástica y un doble marcaje con tetraciclina menor o ausente, aunque en estos casos se observó también un porcentaje elevado de contaminación por aluminio129. En un estudio transversal estos autores encontraron también la asociación entre enfermedad arterial periférica y una reducción significativa en la respuesta esquelética anabólica a la PTH130.
Evidencia adicional de que la hiporrespuesta a PTH es un factor importante en el desarrollo de HPS y/o EOA, deriva de los estudios clínicos que han mostrado que son necesarios niveles elevados de PTH circulante para mantener un remodelado óseo normal16,131,132. De este modo, por ejemplo en pacientes en diálisis, las guías actuales3,133,134 sugieren que el tratamiento debería ser modificado para mantener unos niveles de PTH como mínimo 2 veces por encima del valor superior de la normalidad (siendo mejor en combinación con la fosfatasa alcalina ósea) para evitar una EOA. Asimismo, los pacientes con ERC en estadios previos al inicio de diálisis precisarían unos niveles más elevados de PTH para mantener una superficie osteoblástica normal16, sugiriendo que la diálisis podría mejorar la respuesta a la PTH. De hecho, Torres et al. en este estudio realizado con biopsias óseas en pacientes sin tinción significativa por aluminio parecían demostrarlo16. Aun así, la reversibilidad en diálisis no es una observación uniforme46. Finalmente, la presencia de este complejo multifactorial de hiporrespuesta a la PTH podría explicar la ausencia de asociaciones claras entre los niveles de PTH circulante y las consecuencias clínicas, o sobre mortalidad en pacientes con ERC (normalmente en forma de U-, J- o J invertida, matemáticamente complejas), frente a las asociaciones lineales observadas en el hiperparatiroidismo primario o con los niveles séricos de fosfatasa alcalina135.
CorolarioEn función de las observaciones experimentales descritas previamente, la hiporrespuesta a la PTH es un componente integral del HPS y del complejo CKD-MBD tal y como lo son los niveles elevados de PTH circulante (fig. 3). Las claras diferencias observadas en la respuesta calcémica a la PTH entre pacientes o animales con ERC y controles normales no puede estar tan solo justificada por la presencia de distintos fragmentos inactivos o antagonistas de la PTH, ya que todos los individuos y animales experimentales recibieron el mismo compuesto de PTH de manera constante con resultado dispar (fig. 2). La retención de P, déficit de calcitriol, el FGF23/klotho, esclerostina y/u otros factores urémicos probablemente desempeñan un papel al desensibilizar y/o infrarregular el receptor de la PTH o alterar las vías subsecuentes de transducción de señal.
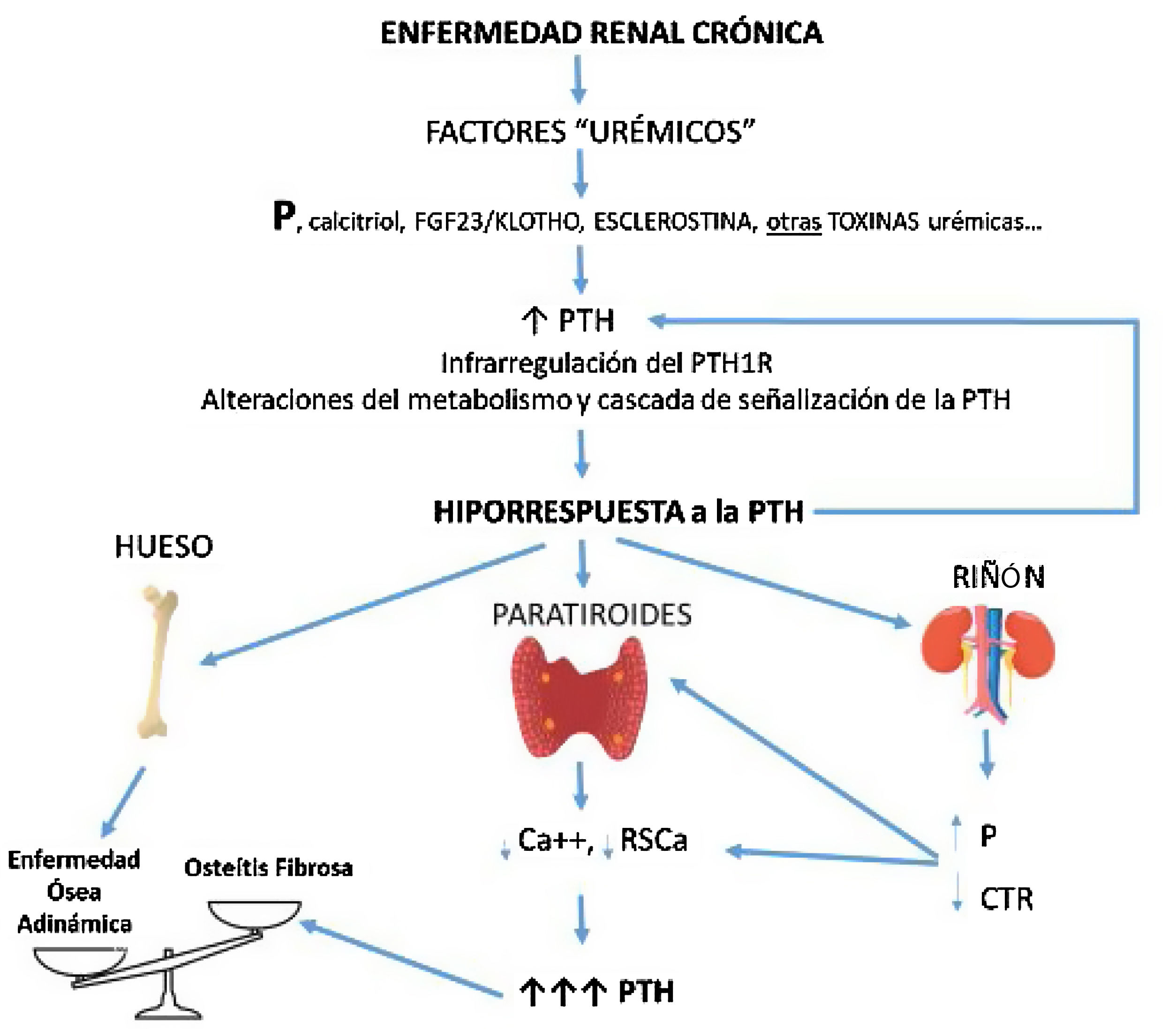
Potencial secuencia temporal y consecuencias de la hiporrespuesta a la PTH en la ERC. Con el desarrollo de la enfermedad renal crónica (i. e. disminución del filtrado glomerular), factores relacionados con la pérdida de función renal (por ejemplo la clásica sobrecarga de fósforo y/o disminución de calcitriol) junto a factores (conocidos o no) asociados a la propia «uremia» producirían a un aumento de PTH que, junto a alteraciones de su metabolismo, interferencias en sus vías de señalización y/o infrarregulación de sus receptores condicionaría incrementos adicionales de PTH debido a su respuesta anormalmente disminuida. Por diferentes mecanismos, a menor respuesta calcémica a la acción de la PTH más hormona es necesaria para restaurar los niveles de calcio a la normalidad (mayor síntesis y secreción de PTH, así como mayor necesidad de proliferación celular). Asimismo, se crearían nuevos círculos viciosos a nivel renal y disbalances en el hueso según predominaran los elementos anabólicos o inhibitorios. La osteítis fibrosa es la expresión ósea del hiperparatiroidismo secundario (enfermedad ósea de alto recambio) y la enfermedad ósea adinámica es resultante de un bajo recambio óseo.
CTR: calcitriol; FGF-23: acrónimo inglés de Fibroblast Growth Factor 23; P: fósforo; PTH1R: receptor de la hormona paratiroidea (PTH); RSCa: receptor sensor de calcio (adaptado de Bover et al.148).
Aunque la resistencia esquelética a la PTH fue inicialmente sugerida como un mecanismo implicado en la patogenia del HPS en la ERC, la «hiporrespuesta» a la PTH también ha sido asociada con el aumento de prevalencia de la EOA (fig. 3). Esta se explicaría también por el incremento de pacientes añosos o diabéticos con ERC, o tratamientos antiparatiroideos excesivamente agresivos. Por lo tanto, los nefrólogos deberíamos tener en cuenta este concepto, ya clásico, de hiporrespuesta a la PTH a la hora de tratar el HPS de nuestros pacientes, siendo importante intentar evitar la completa normalización de los niveles de PTH, tal y como sugieren las guías actuales3,134. Por otra parte, el aumento progresivo y persistente de los niveles de PTH transformará una situación clínica inicialmente adaptativa en una claramente maladaptativa.
Definir un objetivo óptimo de PTH individual es actualmente imposible, pero podría conseguirse en el ámbito poblacional44. Sigue siendo discutible si es mejor esperar a la aparición de HPS severo (grave) antes de empezar el tratamiento antiparatiroideo, como sugieren las últimas guías KDIGO3, o simplemente evitar una normalización de los niveles de PTH, como sugerimos otros134. En cualquier caso parece existir un cierto consenso sobre que el mínimo nivel de PTH con el que se debería llegar a diálisis sería aproximadamente 2X el límite alto de la normalidad para el kit usado3,134. De hecho, ahora podríamos especular que si se corrigieran los factores antes mencionados (retención de P, hipovitaminosis D, infrarregulación del PTHR1, toxinas urémicas, etc.) el hueso y el túbulo renal a lo mejor podrían responder normalmente a los niveles normales de PTH, y esta no tendría que elevarse maladaptativamente.
Probablemente no es plausible una sola recomendación acerca de la PTH para todos los pacientes con ERC (ni antes ni después de haber iniciado diálisis) y deberíamos inclinarnos hacia un manejo más personalizado e individualizado (probablemente compartiendo las decisiones terapéuticas con el paciente)25,136,137, dada la necesidad de tener en cuenta otros factores como la edad, la presencia o ausencia de diabetes, el síndrome metabólico, el síndrome nefrótico, la calcificación vascular, el riesgo de fractura (recientemente incorporado a nuestra práctica clínica) u otros marcadores bioquímicos (p. ej. fosfatasa alcalina). Otros factores o conceptos identificados recientemente y que requieren de futuras investigaciones (toxinas urémicas, FGF23/klotho, Wnt/β-catenina, vías de activación del receptor de activina A tipo 2, «osteólisis osteocítica», etc.) probablemente aportarán nuevas perspectivas138.
Entre tanto, pese a sus limitaciones, ningún biomarcador o estrategia terapéutica ha demostrado ser superior a la PTH, y desde luego necesitamos estudios que permitan mejorar la precisión diagnóstica. La monitorización más frecuente, permitiendo observar tendencias en los niveles de PTH, parece ser hoy la manera adecuada de proceder hasta que estén disponibles nuevos y mejores objetivos moleculares y tratamientos que hayan demostrado eficacia en la práctica clínica.
Finalmente, la resistencia a la acción biológica de otras hormonas, como la insulina, el calcitriol, la hormona del crecimiento y FGF23 es también una característica bien conocida de la ERC139–142, así como la expresión disminuida de diversos receptores (p. ej., RVD, RSCa, FGFR/klotho)65,143–147. De hecho, del mismo modo que la uremia es un trastorno metabólico complejo que implica numerosas moléculas, alteraciones metabólicas y vías aberrantes de señalización en todo el organismo92, la uremia podría ser también considerada como una enfermedad que, por mecanismos no bien conocidos, afecta ampliamente la funcionalidad de diferentes tipos de receptores (uremia, una «enfermedad de receptores»)44,148. De este modo son necesarios estudios que permitan descubrir líneas terapéuticas que podrían ser útiles incluso más allá de la hiporrespuesta a la PTH.
ConclusiónEl HPS es uno de los componentes integrales del complejo CKD-MBD y la hiporrespuesta (o resistencia) a la PTH forma parte de sus mecanismos patogénicos. Esta hiporrespuesta es fruto de múltiples mecanismos complejos (muchos aún no conocidos) y que no son exclusivos de esta hormona. Como consecuencia, un cierto grado de HPS es un mecanismo adaptativo necesario en los pacientes con ERC, y solo futuros estudios a nivel molecular podrán dar respuesta a los mecanismos íntimos de la hiporrespuesta hormonal en la ERC y al descubrimiento de líneas terapéuticas que podrían ser útiles más allá de explicar la hiporrespuesta a la PTH.
Conceptos clave- 1.
El HPS es uno de los componentes integrales de las alteraciones del metabolismo óseo-mineral (CKD-MBD) en la ERC.
- 2.
La PTH es considerada una «toxina urémica» con efectos deletéreos sistémicos más allá del hueso.
- 3.
La hiporrespuesta (o resistencia) a la acción de la PTH hace referencia a la disminución de la respuesta a la acción de la PTH (i. e. respuesta calcémica) que se observa desde fases precoces de la ERC.
- 4.
Hoy en día se prefiere el término «hiporrespuesta» al de «resistencia» a la acción de la PTH, puesto que en la ERC se observa una disminución más que una ausencia de respuesta a la PTH.
- 5.
La hiporrespuesta a la acción de la PTH forma parte de los mecanismos patogénicos del HPS en la ERC.
- 6.
Múltiples mecanismos se asocian a la hiporrespuesta a la PTH, como son la disminución de niveles de calcitriol, retención de P, infrarregulación del PTHR1 renal y óseo, la acumulación de toxinas urémicas y diferentes moléculas como αKlotho, FGF-23, esclerostina o diversos factores que convergen en las vías descendentes de señalización.
- 7.
No es posible identificar un objetivo óptimo de PTH para todos los pacientes con ERC, por lo que se debe optar por un manejo individualizado.
- 8.
No se debería intentar mantener valores normales de PTH en pacientes con ERC, puesto que un cierto grado de HPS es un mecanismo adaptativo necesario.
- 9.
Las guías de práctica clínica subrayan la importancia de tratar tendencias (considerando PTH, Ca y P en su conjunto) y no responder a determinaciones aisladas de PTH.
- 10.
Futuros estudios a nivel molecular podrían dar respuesta a los mecanismos íntimos de la hiporrespuesta a la PTH en la ERC y a la afectación de la función de diferentes receptores en la uremia («uremia, una enfermedad de receptores»).
Los autores no tienen conflicto de intereses directamente relacionado con este tema. J. Bover ha recibido honorarios por conferencias y consultorías de Abbvie, AMGEN, Sanofi y VIFOR-Fresenius-Renal Pharma. P. Ureña ha recibido honorarios por conferencias y consultorías de AMGEN, Astellas, GSK, Hemotech, Leo-Pharma, Sanofi y VIFOR-Fresenius-Renal Pharma. A. Martín-Malo ha recibido honorarios por conferencias y consultorías de VIFOR-Fresenius-Renal Pharma, Medtronic y AstraZeneca.
J. Bover pertenece a la «Red Nacional RedinRen» (RD06/0016/0001 y RD12/0021/0033) y la «Red de Biobancos Nacional Española» (RD09/0076/00064), así como al «Grupo Catalán de Investigación» AGAUR (2009 SGR-1116). A. Torres pertenece también a la «Red Nacional RedinRen» (RD16/0009/0031). Agradecemos al Sr. Ricardo Pellejero su inestimable, constante y entusiasta asistencia bibliográfica y a la Srta. María Fernanda Coll su ayuda en el diseño gráfico.



