







La interleuquina 17A (IL-17A) es una citoquina proinflamatoria producida por células del sistema inmune, sobre todo por los linfocitos Th17 y los linfocitos γδ. En este trabajo, revisamos el papel de IL-17A en la patogenia de la hipertensión y de la lesión en órganos diana. Estudios en ratones han demostrado que la IL-17A aumenta la presión arterial, probablemente por acciones a varios niveles. Además, las concentraciones plasmáticas de IL-17A están ya aumentadas en pacientes con hipertensión arterial ligera o moderada. Estudios preclínicos sobre hipertensión arterial han detectado células productoras de IL-17A en órganos diana, como corazón, vasos y riñón. En pacientes con nefroesclerosis hipertensiva existe infiltración del riñón por linfocitos Th17 y linfocitos γδ que expresan IL-17A. Además, en modelos experimentales de hipertensión el bloqueo de IL-17A, mediante estrategias génicas, o utilizando anticuerpos neutralizantes, disminuye la presión arterial por acciones sobre la pared vascular y el transporte tubular de sodio y disminuye la lesión en órganos diana. En conjunto, los datos presentados en esta revisión sugieren que la IL-17A participa en la regulación de la presión arterial y en la génesis y mantenimiento de la hipertensión arterial, pudiendo constituir una diana terapéutica en el futuro.
Interleukin-17A (IL-17A) is a proinflammatory cytokine produced by cells of the immune system, predominantly Th17 lymphocytes and γδ lymphocytes. In this paper, we review the role of IL-17A in the pathogenesis of hypertension and target organ damage. Studies in mice have shown that IL-17A increases blood pressure, probably by acting on multiple levels. Furthermore, IL-17A plasma concentrations are already elevated in patients with mild or moderate hypertension. Preclinical studies on arterial hypertension have detected IL-17A-producing cells in target organs such as the heart, vessels and kidneys. Patients with hypertensive nephrosclerosis show kidney infiltration by Th17 lymphocytes and γδ lymphocytes that express IL-17A. In addition, in experimental models of hypertension, blocking IL-17A by genetic strategies, or using neutralising antibodies, lowers blood pressure by acting on the vascular wall and tubule sodium transport and reduces damage to target organs. As a whole, the data presented in this review suggest that IL-17A participates in the regulation of blood pressure and in the genesis and maintenance of arterial hypertension, and may constitute a therapeutic target in the future.
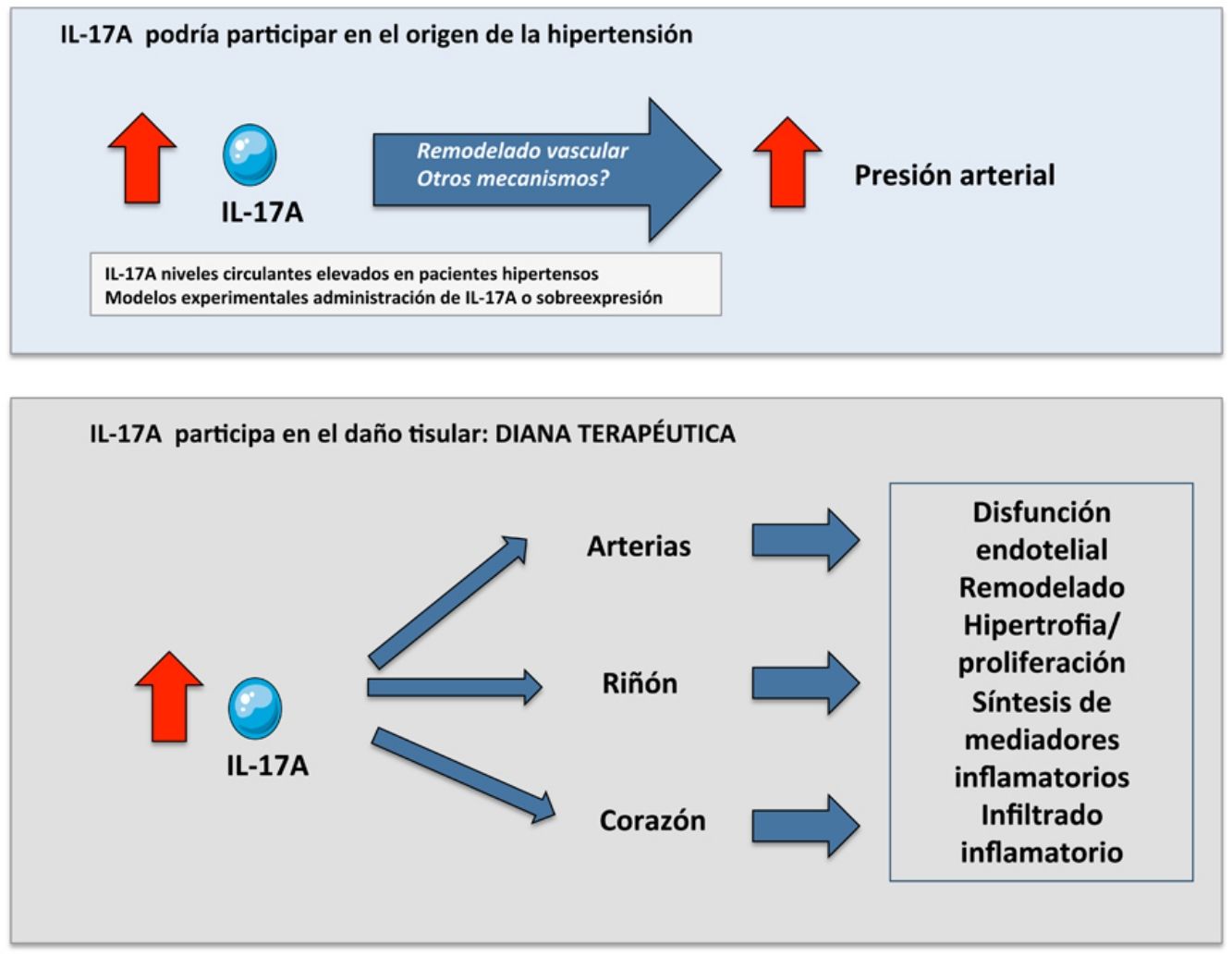
La hipertensión arterial (HTA) se define como una elevación mantenida de la presión arterial por encima de los límites normales, siendo una enfermedad prevalente en nuestra sociedad, asociada con un aumento de la morbimortalidad cardiovascular1. La HTA es una enfermedad silenciosa que puede cursar sin síntomas aparentes, pero puede ocasionar lesiones en órganos diana, como el sistema cardiovascular y los riñones, entre otros. Además, estos órganos están implicados en el control de la presión arterial, lo que contribuye a dificultar aún más la investigación en este campo. La etiología de la HTA esencial continúa sin estar completamente establecida y supone un intenso tema de debate entre la comunidad científica. Recientemente, se ha llamado la atención sobre la participación de la inflamación, los trastornos autoinmunes y el estrés oxidativo en el desarrollo y progresión de la HTA, así como el origen de las lesiones de órganos diana2–7.
En los años 60 se presentaron las primeras evidencias de la participación de la respuesta inflamatoria en la HTA7,8. Apoyando esta hipótesis, los pacientes hipertensos presentan niveles séricos elevados de varias citoquinas proinflamatorias, sugiriendo que la inmunidad innata, tanto celular como humoral, participa en la patogénesis de la HTA9–13. Actualmente, y a pesar de la gran variedad de fármacos disponibles para su tratamiento, el control de la presión arterial no suele ser el óptimo en un número relevante de pacientes, lo que conlleva la existencia de daño orgánico relevante en los órganos diana. Por ello son necesarias nuevas estrategias terapéuticas que mejoren el control de la presión arterial y protejan los órganos diana.
Entre las citoquinas potencialmente implicadas en la génesis y progresión del daño orgánico de la HTA, la interleuquina IL-17A ha adquirido un papel especialmente relevante y es una de las dianas terapéuticas más prometedoras, ampliamente estudiada en enfermedades autoinmunes e inflamatorias crónicas, incluida la propia HTA y la enfermedad renal crónica (ERC)3,6,14.
La presente revisión describe las principales características generales de la citoquina IL-17A y actualiza la información sobre su papel en la patogenia de la HTA y de la lesión de órganos diana.
Características generales de la citoquina IL-17AEn este apartado revisamos las citoquinas IL-17, sus receptores, las células productoras de IL-17A y las funciones de esta citoquina.
Las citoquinas IL-17 y sus receptoresLa familia de citoquinas IL-17 tiene 6 miembros, que incluyen desde IL-17A hasta IL-17F, estando todos ellos implicados en la respuesta frente a infecciones por patógenos y en enfermedades autoinmunes e inflamatorias crónicas15, siendo la citoquina IL-17A la más estudiada dentro del grupo. Esta familia de citoquinas presenta secuencias muy conservadas en mamíferos, de tal forma que entre las isoformas humanas y murinas de IL-17 existe una homología de secuencia entre el 62% en la IL-17A y el 88% en la IL-17B16. Los dos miembros que presentan mayor homología entre sí son la IL-17A y la IL-17F, que pueden formar heterodímeros17. La IL-17A, inicialmente denominada antígeno citotóxico humano asociado a linfocitos T-8, fue aislada por primera vez en 1993 a partir de un hibridoma de células T. Aunque el análisis de su ADNc demostró que presentaba cierta homología con un gen del Herpesvirus samiri, fue clasificada dentro del grupo de las citoquinas debido a que posee una secuencia inestable rica en adenilato-uridilato en la región 3’ UTR y a que es capaz de inducir la síntesis de citoquinas18. A nivel molecular, IL-17A es una glicoproteína de 155 aminoácidos y con un peso molecular de 35kDa, aunque de manera general da lugar a la formación de homodímeros unidos entre sí por un puente disulfuro16,19. Las citoquinas IL-17 activan receptores de la misma familia, los IL-17R que constan de 5 miembros, nombrados desde IL-17RA hasta IL-17RE, y que también pueden formar homo y heterodímeros entre sí. Así, IL-17RA está presente en la mayoría de dímeros que se forman, y en una gran diversidad de tipos celulares, mayoritariamente en células inmunes, pero el resto de receptores son específicos de tipos celulares concretos20. Asimismo, también existen diferencias en cuanto a la afinidad ligando-receptor entre los diferentes miembros, uniéndose lL-17A con mayor afinidad al IL-17RA, mientras que IL-17F se une preferentemente al IL-17RC19,21. Estas diferencias de afinidad podrían ayudar a explicar la gran variabilidad en las respuestas desencadenadas por esta versátil familia de citoquinas.
Células productoras de IL-17ALas primeras células descritas capaces de producir IL-17A fueron los linfocitos Th17 (células CD4+/IL-17A+). De manera general, los linfocitos T CD4+ participan activamente en la respuesta inmune, de tal forma que tras una estimulación antigénica los linfocitos CD4+ naive son activados, y se expanden y diferencian en las distintas subpoblaciones conocidas como T helper (Th)22. Dentro de estas subpoblaciones Th, se incluyen tanto los subtipos efectores Th1, Th2, Th9, Th17, Th22 y Th folicular (Thf), como el subtipo de linfocito T regulador, denominado Treg, ampliando de este modo la visión clásica de únicamente dos subtipos Th (Th1 y Th2)23,24. Cada subtipo Th se clasifica en función del patrón específico de citoquinas que producen y de los diferentes marcadores que expresan23,24. Sin embargo, existe una gran plasticidad entre los diferentes subtipos, existiendo fenotipos celulares intermedios que son objeto de investigación, pero también de controversia25–28. La diferenciación a cada subtipo Th está dirigida por una combinación específica de citoquinas que activan factores de transcripción específicos29–32. En este sentido, la combinación especifica de las citoquinas proinflamatorias IL-1β, IL-6, y/o IL-21 induce la diferenciación de linfocitos T humanos a Th17. Esta diferenciación puede además estar regulada por TGF-β1 y es necesaria la activación tanto del factor RORγt (Retinoid related Orphan Receptor γt) como de la proteína STAT3 (Signal Transducer and Activator of Transcription 3) para inducir la transcripción de genes que darán lugar a la transición fenotípica a Th17, como el gen de IL-23R33–37. Una vez diferenciadas, las células Th17 secretan mayoritariamente IL-17A, pero también producen IL-22, IL-26 e IL-23, que ayudan a la estabilización de la estirpe, o quimioquinas que contienen el motivo C-C, como el ligando quimiocina-20 (CCL-20)29,30,33,38,39 (fig. 1). Con este patrón de secreción, las células Th17 tienen como principal función la defensa frente a patógenos en enfermedades infecciosas, pero también participan en la patogenia de diversas enfermedades inflamatorias y enfermedades autoinmunes, como la artritis reumatoide, enfermedades inflamatorias del intestino, esclerosis múltiple, o enfermedades inflamatorias crónicas, incluidas la aterosclerosis y la HTA40–43. En este sentido, varios factores relacionados con el daño cardiovascular y renal, como la citoquina IL-6, la Angiotensina II (Ang II), el TGF-β1, o el CTGF/CCN2 (connective tissue growth factor) participan en la generación de células Th1744–46. La IL-17A es producida por otras células además de las Th17, incluidos los linfocitos γδ (fig. 1), y probablemente también por neutrófilos, células invariant natural killer T cells, células CD8+, células linfoides innatas y mastocitos, aunque en algunas de estas células existe controversia respecto a si solo son capaces de almacenar la IL-17A pero no de producirla, como podría ser el caso de los mastocitos30,37,47,48.
Efectos celulares de IL-17A
IL-17A genera respuestas proinflamatorias que pueden variar sustancialmente en función del tipo celular y las condiciones patológicas9,10,11,13,49–54. Una de las primeras evidencias de la implicación de IL-17A en la respuesta inflamatoria mostró que en sinoviocitos cultivados de pacientes con artritis reumatoide, IL-17A aumentaba los niveles de IL-6 e IL-854. Posteriormente, se observó que IL-17A inducía respuestas en diversas células inmunes, regulando varias funciones, como la quimiotaxis de monocitos, y el aumento de la producción de mediadores proinflamatorios, contribuyendo así a amplificar la respuesta inflamatoria en los tejidos dañados11,52,53,55,56 (fig. 2). Las acciones de IL-17A en las células del sistema inmune no están del todo esclarecidas, pudiendo además participar en el cambio de fenotipos de macrófagos57, e inducir la degradación de matriz extracelular por neutrófilos, mediante la regulación de metaloproteinasas (MMPs)58.
IL-17A en hipertensión en células inmunes y en la respuesta inflamatoria tisular.")
Los pacientes hipertensos presentan niveles plasmáticos elevados de citoquinas proinflamatorias, incluyendo la IL-17A59–61. También se ha observado una clara asociación entre los niveles séricos elevados de IL-17A y un estado de pre-HTA41,61,62. En esta misma línea, los niveles circulantes de IL-17A también están incrementados en diversas patologías que cursan con HTA asociada a enfermedades autoinmunes, incluyendo lupus eritematoso sistémico, así como en la preeclampsia y el rechazo crónico del trasplante2,10,62,63,64. Además, el número elevado de células T CD4+ circulantes productoras de IL-17A también está aumentado en pacientes hipertensos5, apoyando aún más la hipótesis de la participación de IL-17A en el desarrollo y la progresión de la HTA. Aunque la HTA no ocasiona hipernatremia, incrementos leves en la concentración de sodio en pacientes hipertensos pueden dar lugar a la polarización de células T indiferenciadas a células Th17, favoreciendo la autoinmunidad, la inflamación y la sobrerregulación del cotransportador Na-K-2CL (NaKCCl)7. En pacientes con nefroesclerosis hipertensiva localizamos por primera vez células productoras de IL-17A en el riñón, que fueron identificadas como células Th17 (CD4+/IL-17A+) y linfocitos γδ65.
Estudios preclínicos de IL-17A en hipertensión y en lesión en los órganos dianaA nivel experimental, existen numerosos datos apoyando el papel de los linfocitos T, en concreto de las células Th17 y su citoquina efectora IL-17A, en la regulación de la HTA2. Las primeras evidencias surgieron en ratones deficientes en linfocitos T y B (ratones RAG1−/−), que estaban protegidos de la HTA y de las lesiones vasculares inducidas por la administración sistémica de Ang II. Experimentos de transferencia de células T y células B demostraron que solo las células T restauran los efectos de la Ang II en los ratones RAG1−/−66. Otros de los procesos patológicos en los que se ha sugerido la participación de las células Th17 en la hipertensión pulmonar asociada a hipoxia67 y la HTA causada por inmunosupresores inhibidores de la calcineurina, como ciclosporina A y tacrolimus68.
Otros estudios preclínicos han mostrado la presencia de células que expresan IL-17A en los tejidos dañados por HTA, incluyendo el sistema cardiovascular y los riñones2,41,69. De este modo, la administración de Ang II a ratones aumenta los linfocitos T infiltrantes que expresan IL-17A tanto en la adventicia como en la grasa periadventicial de la aorta41. Posteriormente, otros estudios identificaron los linfocitos Th17 y linfocitos γδ, como las células productoras de IL-17A en los riñones y aortas de ratones infundidos con Ang II69.
Los linfocitos γδ están implicados en la respuesta inmune frente a hongos y patógenos, así como en enfermedades autoinmunes70,71. Se trata de células T no convencionales, que reconocen muchos microorganismos y transforman células huésped, actuando como la primera línea de defensa en tejidos periféricos72,73. La IL-17A producida por estas células participa principalmente en la inmunidad antifúngica, como la respuesta frente a infección por Candida albicans70; y en las etapas iniciales de patologías autoinmunes74. Los linfocitos γδ productores de IL-17A se generan en la periferia, pudiendo ser reclutados y acumulados en tejidos dañados, como la piel, contribuyendo a la inflamación sostenida, como se ha observado en la psoriasis72. Los linfocitos γδ son también la principal fuente de IL-17A en los corazones hipertróficos de ratones infundidos con Ang II75. En este sentido, se han observado linfocitos γδ que expresan IL-17A en el riñón de pacientes hipertensos65.
Estos datos sugieren la posibilidad de un nuevo abordaje terapéutico basado en la inhibición de estas células para hacer frente al daño tisular asociado a HTA.
Modulación de IL-17A en hipertensión experimentalDiversos estudios experimentales avalan la hipótesis de la participación de IL-17A en la patogenia de la HTA4,41,69,76. En este sentido, el bloqueo de IL-17A mediante la deleción génica de la citoquina o de sus receptores, o mediante anticuerpos neutralizantes frente a IL-17A, disminuye la presión arterial en modelos experimentales de HTA41,69. Sin embargo, la deficiencia del eje IL-17/IL-23 no modificó la HTA inducida por la combinación de DOCA (del inglés deoxycorticosterone acetate) y Ang II77. Otros trabajos preclínicos demostraron que IL-17A puede aumentar directamente la presión arterial, como se ha observado en el ratón transgénico que sobreexpresa IL-17A específicamente en queratinocitos78, o por administración de IL-17A intraperitoneal (1μg/día)76 o por vía sistémica65,79. En estos dos últimos estudios la dosis de IL-17A administrada indujo niveles séricos similares a los detectados en sujetos con cifras de presión arterial en el rango de 120-130/80-89mmHg62, valores que actualmente se considera que exceden los niveles óptimos80. Otros estudios preclínicos observaron una asociación entre el aumento de la presión arterial inducido por IL-17A y la aparición de lesiones en órganos diana tales como disfunción endotelial, cambios estructurales y funcionales a nivel vascular y cardíaco o una respuesta inflamatoria a nivel cardíaco, vascular y renal67,76,79. En su conjunto, estos datos sugirieren que niveles circulantes elevados de IL-17A podrían contribuir tanto al desarrollo de la HTA como a la inducción de lesión de órgano diana y postulan esta citoquina como potencial biomarcador y/o diana terapéutica.
Mecanismos moleculares regulados por IL-17A a nivel vascularEntre los mecanismos que podrían contribuir al remodelado arterial asociado a HTA se encuentran la inflamación, el número y tamaño celular (proliferación, apoptosis y/o hipertrofia), cambios en el fenotipo celular y modificaciones en la composición de la matriz extracelular que pueden dar lugar a fibrosis vascular81 (fig. 3). IL-17A regula algunos de estos mecanismos a nivel vascular, a través de los cuales podría regular la HTA y el daño vascular asociado.
IL-17A y la respuesta inflamatoria en el daño vascular
La inflamación se define como una respuesta no específica que sufre un tejido ante una agresión producida por diferentes factores mecánicos, químicos o biológicos, y que tiene como finalidad la supresión del agente causante del daño y la reparación del tejido dañado82. Estudios epidemiológicos han demostrado que existe una importante conexión entre diferentes enfermedades inflamatorias crónicas y enfermedades cardiovasculares. En este sentido, la incidencia de infarto de miocardio está aumentada en pacientes con artritis reumatoide, lupus eritematoso sistémico o psoriasis83. Estos datos contribuyen a resaltar la importancia que tiene la respuesta inflamatoria en el desarrollo y la progresión de diversas patologías vasculares, como la vasculitis, los aneurismas, la HTA o la aterosclerosis83,84.
Estudios recientes han evaluado el papel de la inflamación vascular y de citoquinas y quimioquinas concretas (vg MCP-1, IL-8, IP-10 e IL-17A) en la aparición y desarrollo de la disfunción endotelial asociada a HTA85. IL-17A es una citoquina clave en la respuesta inflamatoria, incluido en el sistema cardiovascular (fig. 2). De hecho, la administración de IL-17A aumenta la expresión de múltiples genes proinflamatorios, incluidos mcp-1 e il-6, en células musculares lisas vasculares (CMLVs) murinas en cultivo79. Del mismo modo, la IL-17A potencia el efecto proinflamatorio de otras citoquinas en células endoteliales, CMLVs y macrófagos86. Respaldando estos hallazgos, la deficiencia genética de IL-17A disminuyó el infiltrado inflamatorio en la pared aórtica murina a niveles normales41.
Los principales mecanismos proinflamatorios activados por IL-17A incluyen la producción de especies reactivas de oxígeno (ROS), de óxido nítrico (NO), la activación del factor de transcripción factor nuclear-kappaB (NF-κB), así como la regulación de diversas cascadas de señalización asociadas a proteínas quinasas, como RhoA/Rho-quinasa, proteínas quinasas activadas por mitógeno (MAPKs) o Akt2,14,52,53,55,65,76,78,87 (fig. 4). NF-κB es una molécula clave en la regulación de la respuesta inflamatoria, y también en el remodelado vascular88, que contribuye al desarrollo y la regulación del daño cardiovascular en la HTA, la aterosclerosis, la hipertrofia cardíaca, el infarto de miocardio o la formación de aneurismas88–92. En células endoteliales de cordón umbilical humano (HUVEC) en cocultivo con células T (células Jurkat) demostraron que IL-17A facilita la inflamación endotelial, al inducir la expresión de las moléculas de adhesión ICAM-1 y VCAM-1 y las quimiocinas CCL-20 y CXCR-4 en las células endoteliales de manera dependiente de la fosforilación de ERK1/2, y por tanto promueve la adherencia de las células T a las HUVEC87 (fig. 4). Un estudio reciente ha demostrado que la sobreexpresión de IL-17A en células T en ratones provoca un aumento en la producción de ROS en células circulantes, disfunción vascular y fibrosis perivascular comparado con ratones control93.

Últimamente se ha demostrado que el receptor TLR-4 (del inglés Toll-like receptor 4) participa en la HTA94 y en la regulación de la inflamación renal experimental95,96. En este sentido, los ratones deficientes en TLR-4 están parcialmente protegidos de la HTA y de la sobreproducción de ROS, la infiltración de macrófagos y la expresión de MCP-1 a nivel renal inducida por Ang II97. Curiosamente, el bloqueo de la IL-17A disminuyó considerablemente la expresión del mRNA de TLR-4 en riñón inducida por la infusión de Ang II, lo que sugiere una interconexión entre IL-17A y TLR-4 en la regulación de la HTA y lesión renal desencadenadas por Ang II65.
IL-17A y disfunción endotelialIL-17A induce disfunción endotelial y estrés oxidativo, a través de la vía de señalización intracelular RhoA/Rho-quinasa (ROCK), mediante fosforilación del residuo de treonina 495 de la óxido nítrico sintasa endotelial (de sus siglas en inglés eNOS Thr495) en aorta de ratón76,78,98,99 (fig. 4). Asimismo, los ratones deficientes de IL-17A están protegidos de la disfunción endotelial inducida por Ang II, evaluada como vasodilatación dependiente del endotelio, la contracción inducida por fenilefrina y la producción de ROS41. La vía RhoA/Rho-quinasa y la regulación del NO contribuyen a la HTA mediada por IL-17A, así como a la preeclampsia y a enfermedades autoinmunes asociadas con HTA, como el lupus eritematoso sistémico y el rechazo crónico del aloinjerto76. La sobreexpresión condicional a nivel dérmico de IL-17A en queratinocitos causa una disfunción endotelial sistémica, asociada con una mayor formación de ROS y leucocitos inflamatorios circulantes CD11b+, aumento de la presión arterial sistólica, hipertrofia ventricular izquierda y reducción de la supervivencia en comparación con los controles78. En células endoteliales, la IL-17A activa la expresión de eNOS y ciclooxigenasa-2 (COX-2) y esto se asocia a angiogénesis, determinada como proliferación, apoptosis, migración y tubulogénesis100.
IL-17A y matriz extracelular vascularLa rigidez arterial y la acumulación de matriz extracelular (MEC) pueden contribuir al remodelado vascular y ser la causa o la consecuencia de la HTA, siendo este tema aún muy controvertido101. Los estudios sobre el efecto de la modulación de IL-17A en la fibrosis vascular proporcionaron resultados variables. La administración de IL-17A en arterias mesentéricas de resistencia (AMRs) in vivo aumentó la rigidez vascular intrínseca (independiente de la geometría del vaso), lo cual podría contribuir al aumento en la presión arterial, pero no modificó la estructura tridimensional de la elastina ni los niveles de colágeno, principales componentes de la MEC en las AMRs79. En esta misma línea, y apoyando la ausencia de efecto profibrótico de IL-17A a nivel vascular, en la estenosis experimental por ligadura parcial de arteria carótida caracterizada por remodelado vascular y el aumento de proteínas de MEC, la deleción génica de IL-17A no modificó el porcentaje de estenosis pero sí redujo el remodelado exterior98. Además, la deleción de IL-17A en la aterosclerosis experimental en ratones deficientes en Apolipoproteína E (ApoE) no modificó el área de las placas ateroscleróticas ni los niveles de proteínas de MEC, tales como elastina y colágeno98. Con respecto a la rigidez vascular inducida por la administración sistémica de Ang II, el bloqueo de IL-17A no mejoró la rigidez en las AMRs tras 2 semanas de infusión79, pero la deleción génica de IL-17A revirtió los cambios en la rigidez inducidos por Ang II en la aorta a tiempos más largos de 4 semanas102. A nivel celular, IL-17A no modificó la expresión génica de factores profibróticos ni de componentes de la MEC en CMLVs cultivadas79 (fig. 4). Sin embargo, estudios en cultivos de fibroblastos de aorta mostraron que IL-17A induce la expresión del gen de colageno 3a1102. Estudios en ratones deficientes en células γδT o anticuerpos frente a γδT demuestran que se bloquea la producción de IL-17A en el corazón de ratones infundidos con Ang II asociado a una menor fibrosis cardíaca75. En resumen, diversos estudios han demostrado efectos contradictorios de IL-17A sobre la fibrosis, sugiriendo que esta citoquina presenta acciones diferentes en función del tipo celular y la patología implicada103.
IL-17A y el crecimiento y fenotipo celularIL-17A regula el crecimiento celular en células cultivadas, aunque el efecto depende del tipo celular. De este modo, mientras que en cardiomiocitos IL-17A induce apoptosis104, en CMLVs79,105, fibroblastos106 y células endoteliales107 se ha postulado como un factor proliferativo y migratorio. En un contexto de microambiente tumoral, IL-17A desencadenó proliferación celular y transición epitelio-mesénquima (TEM) evidenciado fundamentalmente por la inhibición de marcadores epiteliales, como E-cadherina, y el aumento de marcadores mesenquimales, como la vimentina108. En este sentido, algunos estudios preclínicos, en líneas celulares humanas de cáncer de próstata109 y en ratones tratados con un anticuerpo neutralizante anti-IL-17A110, demostraron la participación crucial de IL-17A en el desarrollo de TEM. Recientemente, se ha involucrado a la IL-17A producida por neutrófilos en la progresión del cáncer gástrico al inducir TEM por activación de la vía JAK/STAT3111. La participación activa de IL-17A en la modulación del ciclo celular en un contexto tumoral sugiere que también podría desencadenar efectos en un ambiente fisiológico. En células tubulares cultivadas se ha confirmado la participación de IL-17A en la regulación del fenotipo celular, concretamente a la TEM, que puede contribuir a la fibrosis tubulointersticial109,112. A nivel vascular, el remodelado implica cambios en las CMLVs con una transición fenotípica desde el tipo celular contráctil clásico a un fenotipo poco contráctil, conocido como de tipo sintético. Estas transformaciones celulares están asociadas a cambios en los niveles de una serie de proteínas, tanto celulares como del secretoma, que son características de cada fenotipo101,113. En estudios in vivo, IL-17A indujo cambios en la expresión de calponina en CMLVs de las AMRs, lo que refleja un cambio en el fenotipo celular79 (fig. 5). Aunque clásicamente se ha considerado que la rigidez arterial es consecuencia del aumento de MEC, principalmente por acumulación de colágeno, recientemente se ha demostrado que la rigidez también puede aparecer en ausencia de fibrosis, estando en este caso regulada por proteínas que controlan la contractilidad de las CMLVs y las interacciones célula-MEC entre las que se encuentra la calponina101. La calponina es una proteína de unión a actina que regula las funciones contráctiles y la estabilización de las fibras de estrés en CMLVs114 y que disminuye en varios modelos de remodelado vascular hipertrófico115,116. La disminución de los niveles de calponina en las células vasculares de las AMRs inducida por IL-17A podría contribuir a aumentar la rigidez vascular y la presión arterial (fig. 4).

Por otro lado, es importante señalar el papel que desempeña la IL-17A en promover la senescencia de células endoteliales cultivadas117 (fig. 4) e in vivo, según resultados obtenidos en ratones knockout para IL-17A o para su receptor118. Así, recientemente, se ha descrito que un anticuerpo neutralizante para IL-17 redujo la expresión del marcador de senescencia Cdkn1a119.
Potenciales mecanismos de IL-17A en el control de la presión arterial y en la lesión órgano diana: acciones a nivel vascularLos mecanismos que regulan la presión arterial son complejos, e implican la participación de diferentes órganos y sistemas. Uno de los mecanismos que contribuyen a la HTA es la aparición de cambios estructurales y funcionales en las arterias (proceso conocido como remodelado vascular), como la reducción del diámetro del lumen del vaso o el incremento de la relación entre la capa media y el lumen, generando un aumento en la presión que ejerce la sangre sobre la pared de los vasos. El remodelado vascular depende de procesos celulares como el crecimiento celular, la hipertrofia de las CMLVs o la sobreproducción de proteínas de MEC, como colágeno y fibronectina120–122. En concreto, el remodelado de las arterias de pequeño calibre o arteriolas participa en la patogenia de la HTA. El remodelado vascular de las AMRs en pacientes con HTA esencial se caracteriza por un aumento del grosor de la pared vascular, de la relación media/lumen y de la rigidez de la pared. Estos cambios aumentan la resistencia vascular periférica aumentando la presión arterial123–125. La mayor parte de los estudios preclínicos que han evaluado el papel de IL-17A en HTA experimental se han centrado en el estudio de cambios estructurales y funcionales en la aorta41,76,78. Sin embargo, los cambios en la aorta no serían suficientes para explicar el aumento de presión arterial, lo que sugiere que podrían ser consecuencia de la propia HTA. En este sentido, la IL-17A participa en el remodelado vascular de las AMRs sugiriendo que esta citoquina podría ser un factor causante de la HTA79. En ratones, la administración sistémica de IL-17A para aumentar los niveles circulantes de IL-17A a valores similares a los observados en pacientes prehipertensos provoca un aumento en la presión arterial asociado a cambios estructurales y funcionales de las AMRs, caracterizados por un remodelado hipertrófico hacia el interior y un aumento de la rigidez arterial79. Además, una terapia combinada de hidralacina e hidroclorotiazida, administrada cuando IL-17A ya había inducido un aumento en la presión arterial, fue capaz disminuir la presión arterial, pero no de revertir los cambios en las propiedades mecánicas y estructurales de las AMRs inducidos por IL-17A, sugiriendo un efecto directo de IL-17A sobre las AMRs. Apoyando esta hipótesis, demostramos que un anticuerpo neutralizante de IL-17A normalizó el remodelado vascular en las AMRs inducido por la administración sistémica de Ang II79. Estos estudios preclínicos sugieren que los cambios inducidos por la IL-17A en las arterias pequeñas podrían ser responsables, al menos en parte, del aumento de la presión arterial, demostrando un nuevo mecanismo con el que IL-17A podría contribuir al desarrollo de la HTA (fig. 5).
Otros mecanismos de control de la presión arterial por IL-17AEl riñón es un órgano clave en la regulación de la presión arterial, y es también una diana de las acciones de IL-1777,126. Desde el punto de vista fisiológico, únicamente la insuficiente eliminación de sal y agua por el riñón puede ya aumentar la presión arterial de forma sostenida127. Diversas citoquinas modulan el balance hídrico y salino por alteración del tono simpático, provocando disfunción endotelial con efectos secundarios sobre el flujo sanguíneo renal o bien modulando el transporte de sodio a lo largo de la nefrona128. En este sentido, los linfocitos B y las células dendríticas pueden alterar la presión arterial indirectamente al facilitar la activación de las células T. Citoquinas proinflamatorias de células Th1, Th17 y macrófagos, como TGF-β1, TNF-α, IFN-γ, IL-1β y IL-17A, aumentan la presión arterial y/o el daño renal129. A nivel renal, IL-17A aumenta la reabsorción de sodio a través del intercambiador de sodio-protones tipo 3 (NHE-3) en el túbulo proximal y del cotransportador de sodio-cloro (NCC) en el túbulo contorneado distal, contribuyendo a la HTA126. Asimismo, la deficiencia de IL-17A suprimió la activación de los transportadores del túbulo distal, en concreto del cotransportador sodio-potasio y del canal epitelial de sodio, y disminuyó el daño renal inducido por Ang II126. En ratones, la administración de Ang II produjo HTA y redujo la capacidad para excretar una sobrecarga de salino, y de activación de diversos transportadores de sodio de los túbulos proximales y distales. Sin embargo, la respuesta hipertensiva a la administración sistémica de Ang II fue limitada en los ratones deficientes en IL-17A, que conservaron la capacidad de excreción renal de una sobrecarga de sodio, fundamentalmente por una menor actividad de los transportadores de sodio del túbulo proximal130, lo que sugiere que IL-17A interfiere con la natriuresis inducida por un aumento de la presión arterial. Todos estos datos demuestran que IL-17A regula la presión arterial por mecanismos complejos que actúan en diferentes tejidos y sistemas.
PerspectivasEn resumen, en este manuscrito hemos revisado datos actuales implicando a la IL-17A como una citoquina relevante en la patogenia de la HTA y en el daño renal y cardiovascular. En modelos preclínicos de HTA y de enfermedad renal, la inhibición de IL-17A mejoró las lesiones renales, incluso administrado de forma terapéutica, como se ha observado en un modelo de nefropatía diabética131,132. Estos datos apoyan la hipótesis de que la IL-17A es un efector del daño tisular, también en HTA, y por tanto podría considerarse como una potencial diana terapéutica en esta situación clínica. Actualmente están en desarrollo varios ensayos clínicos bloqueando IL-17A con resultados prometedores en trastornos proliferativos hematológicos y en enfermedades autoinmunes132. Una vez demostrada su seguridad clínica, podrían evaluarse como fármacos para tratar algunos casos de HTA resistente y/o prevenir la lesión de órgano diana por HTA.
FinanciaciónEl presente trabajo ha sido financiado por la Sociedad Española de Nefrología y por ayudas del Instituto de Salud Carlos III (ISCIII) y Fondos FEDER European Union (PI17/00119 y Red de Investigación Renal (REDINREN): RD16/0009, a M.R-O, RS, PI17/01495 to JE), Comunidad de Madrid («NOVELREN» B2017/BMD-3751 to M.R-O); the José Castillejo grant (CAS19/00133 to R.R.R-D); «Juan de la Cierva Incorporacion» del Ministerio de Economía, Industria y Competitividad (MINECO) para SR-M (IJC2018-035187-I); «Convocatoria Dinamización Europa Investigación 2019» MINECO (EIN2019-103294 a M.R-O y SR-M); IMPROVE-PD project («Identification and Management of Patients at Risk–Outcome and Vascular Events in Peritoneal Dialysis») del Horizon 2020 Marie Skłodowska-Curie Grant Agreement No. 812699 a M.R.O, y Fondecyt Chile 1160465.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



