


La hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (HFHN) es un trastorno tubular que se caracteriza por la excreción urinaria excesiva de magnesio y calcio y por el deterioro progresivo de la función renal, con evolución a enfermedad renal crónica en relación con los depósitos de calcio en el parénquima renal1. Se trasmite con herencia autosómica recesiva, asociada a la mutación en los genes CLDN16 y 19, que codifican la claudina-16 y 19, que intervienen en el transporte paracelular del calcio y del magnesio a través de las tight junctions, o zonas de unión intercelulares de la membrana apical en el segmento grueso de la rama ascendente de Henle2-5.
El cuadro clínico incluye síntomas directamente relacionados con la tubulopatía, como poliuria, polidipsia, presencia de litiasis, nefrocalcinosis e infecciones de repetición. Por otro lado, se han asociado otros síntomas, como alteraciones neuromusculares y alteraciones oculares3-6.
Presentamos el caso de un varón de 29 años con antecedentes desde la infancia de hipoacusia neurosensorial moderada en seguimiento por Otorrinolaringología, además de nistagmo horizontal, miopía y astigmatismo en seguimiento por Oftalmología, y trastorno paranoide con baja adherencia terapéutica en seguimiento por Psiquiatría.
Al estudiar sus antecedentes familiares, tenía dos hermanos sanos, y además existía una relación consanguínea colateral, al ser sus padres primos.
En marzo de 2020 es derivado a consultas de Nefrología por deterioro progresivo de la función de renal, con creatinina 2,03mg/dl y FGe 41ml/min, y leucocituria estéril; ya presentaba en mayo de 2017 un filtrado glomerular estimado (FGe) de 52ml/min (Cr1,76mg/dl). Refería encontrarse clínicamente asintomático, aunque mantenía poliuria de unos 2-3l (V/GFR=1,88) y polidipsia habitual, no apreciada como excesiva por el paciente.
Ecográficamente mostraba riñones de aspecto crónico disminuidos de tamaño. A nivel medular se observó material hiperecogénico compatible con nefrocalcinosis (fig. 1).

Se citó para revisión en consultas de Nefrología, ampliando el estudio. Sin embargo, el paciente acude a Urgencias por calambres y sensación de «hormigueo», de 3semanas de evolución, con empeoramiento en los últimos días, quedando ingresado en Nefrología para tratamiento sintomático y completar el estudio.
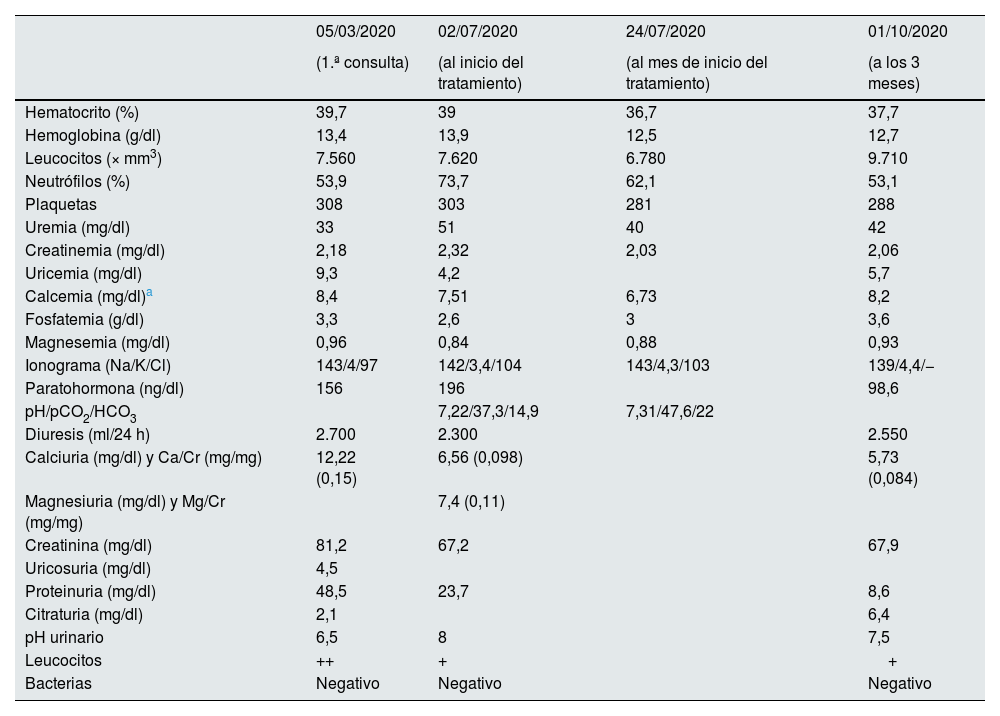
Analíticamente se objetivó deterioro de la función renal, con hipocalcemia (7,7mg/dl), hipomagnesemia (0,96mg/dl), acidosis metabólica normoclorémica, GAP elevada, hiperuricemia e hiperparatiroidismo (tabla 1).
Evolución de datos bioquímicos en sangre y orina de primera consulta de Nefrología, al diagnóstico, al mes y a los 3 meses de tratamiento
| 05/03/2020 | 02/07/2020 | 24/07/2020 | 01/10/2020 | |
|---|---|---|---|---|
| (1.ª consulta) | (al inicio del tratamiento) | (al mes de inicio del tratamiento) | (a los 3 meses) | |
| Hematocrito (%) | 39,7 | 39 | 36,7 | 37,7 |
| Hemoglobina (g/dl) | 13,4 | 13,9 | 12,5 | 12,7 |
| Leucocitos (× mm3) | 7.560 | 7.620 | 6.780 | 9.710 |
| Neutrófilos (%) | 53,9 | 73,7 | 62,1 | 53,1 |
| Plaquetas | 308 | 303 | 281 | 288 |
| Uremia (mg/dl) | 33 | 51 | 40 | 42 |
| Creatinemia (mg/dl) | 2,18 | 2,32 | 2,03 | 2,06 |
| Uricemia (mg/dl) | 9,3 | 4,2 | 5,7 | |
| Calcemia (mg/dl)a | 8,4 | 7,51 | 6,73 | 8,2 |
| Fosfatemia (g/dl) | 3,3 | 2,6 | 3 | 3,6 |
| Magnesemia (mg/dl) | 0,96 | 0,84 | 0,88 | 0,93 |
| Ionograma (Na/K/Cl) | 143/4/97 | 142/3,4/104 | 143/4,3/103 | 139/4,4/− |
| Paratohormona (ng/dl) | 156 | 196 | 98,6 | |
| pH/pCO2/HCO3 | 7,22/37,3/14,9 | 7,31/47,6/22 | ||
| Diuresis (ml/24 h) | 2.700 | 2.300 | 2.550 | |
| Calciuria (mg/dl) y Ca/Cr (mg/mg) | 12,22 (0,15) | 6,56 (0,098) | 5,73 (0,084) | |
| Magnesiuria (mg/dl) y Mg/Cr (mg/mg) | 7,4 (0,11) | |||
| Creatinina (mg/dl) | 81,2 | 67,2 | 67,9 | |
| Uricosuria (mg/dl) | 4,5 | |||
| Proteinuria (mg/dl) | 48,5 | 23,7 | 8,6 | |
| Citraturia (mg/dl) | 2,1 | 6,4 | ||
| pH urinario | 6,5 | 8 | 7,5 | |
| Leucocitos | ++ | + | + | |
| Bacterias | Negativo | Negativo | Negativo |
En orina presentaba pH de 8, con excreción elevada de magnesio y disminuida de ácido úrico. La excreción urinaria de calcio se encontraba dentro de los niveles de normalidad, aunque anormalmente elevada en relación con los niveles en sangre (tabla 1). La oxaluria era normal.
Los hallazgos orientaron al diagnóstico de un trastorno tubular con pérdida urinaria excesiva de magnesio y de calcio. Tanto la historia familiar, con datos de consanguinidad, y la asociación desde la infancia de alteraciones oculares, eran compatibles con HFHN.
Por todo ello, se solicitó un análisis genético, que detectó en homocigosis un cambio en el gen CLDN19 (c.59G>A (p.G20D), descrito en la bibliografía como «mutación española/hispánica o fundadora». Esta da lugar al cambio de glicina por ácido aspártico en la posición 20 de la claudina-19, que altera el péptido señal y hace que la proteína mutante quede retenida dentro de la célula y no llegue a su localización en la membrana celular1-2.
Se pautó tratamiento con alopurinol, carbonato cálcico, citrato potásico, bicarbonato sódico y lactato magnésico, con buena evolución clínica, aunque con difícil control de la magnesemia y la calcemia. En las revisiones en consultas en los meses posteriores no se observó mayor deterioro de función renal.
La asociación de las alteraciones oculares en la HFHN, como el nistagmo, la miopía y el coloboma, ha sido bien definida y estudiada en la bibliografía, y se encuentra estrechamente relacionada con la mutación del gen CLDN19 (hasta en el 90% de los pacientes), pero no así con el gen CLDN165-6.
Sin embargo, se desconoce si pudiera existir una asociación entre la HFHN y las alteraciones auditivas. Diversos estudios han demostrado que la hipomagnesemia incrementa la susceptibilidad para el desarrollo de hipoacusia neurosensorial. Se conoce que el magnesio tiene un importante efecto neuroprotector y vasodilatador a nivel auditivo7-8. Los niveles persistentemente bajos de magnesio en estos pacientes con HFHN podrían predisponerlos a sufrir una pérdida auditiva neurosensorial o a padecer sordera súbita, como en el caso clínico.
Por otro lado, sí se han relacionado mutaciones en otras claudinas con alteraciones auditivas, como la mutación del gen CLDN14 en la sordera autosómica recesiva DFNB299. Las claudinas 14 y 19 se encuentran presentes en el epitelio de las células ciliadas de la cóclea, y en modelos murinos se ha descrito su expresión en el desarrollo posnatal10. Sin embargo, hasta el momento se desconoce si mutaciones en el gen CLDN19 podrían producir alteraciones auditivas, asociadas o no a la HFHN.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesNinguno.



